




优先发表栏目展示本刊经同行评议确定正式录用的文章,这些文章目前处在编校过程,尚未确定卷期及页码,但可以根据DOI进行引用。
当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024012
摘要:
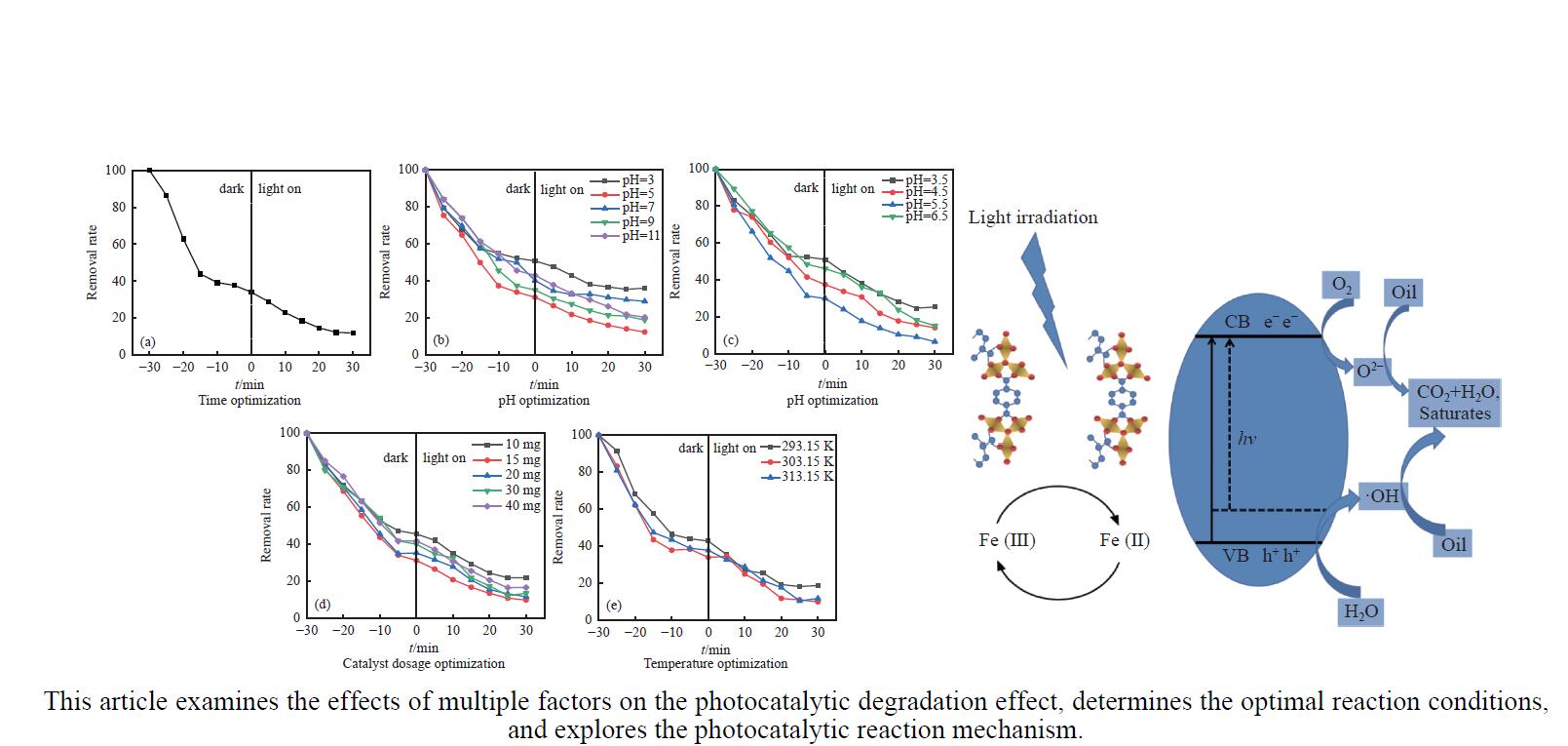
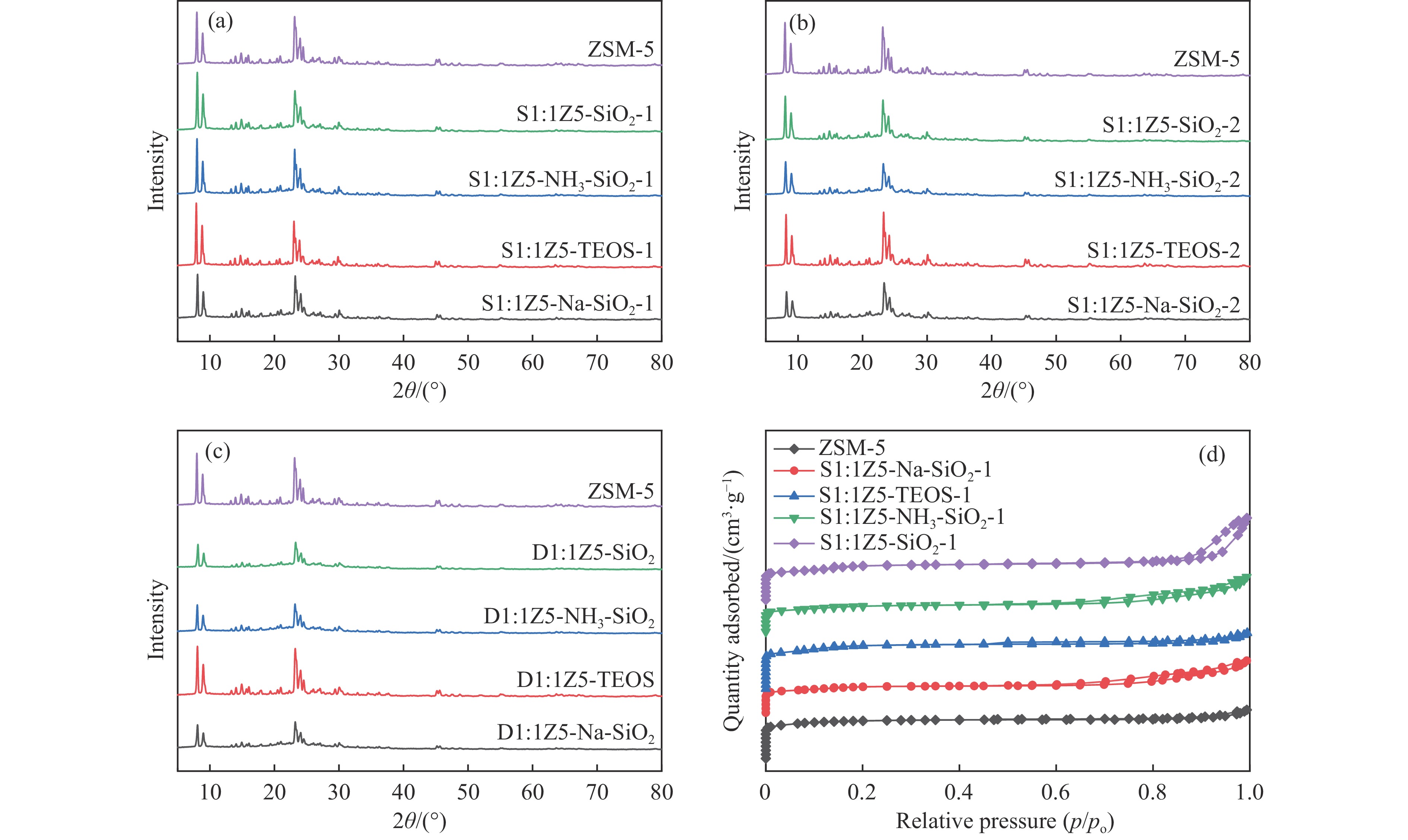
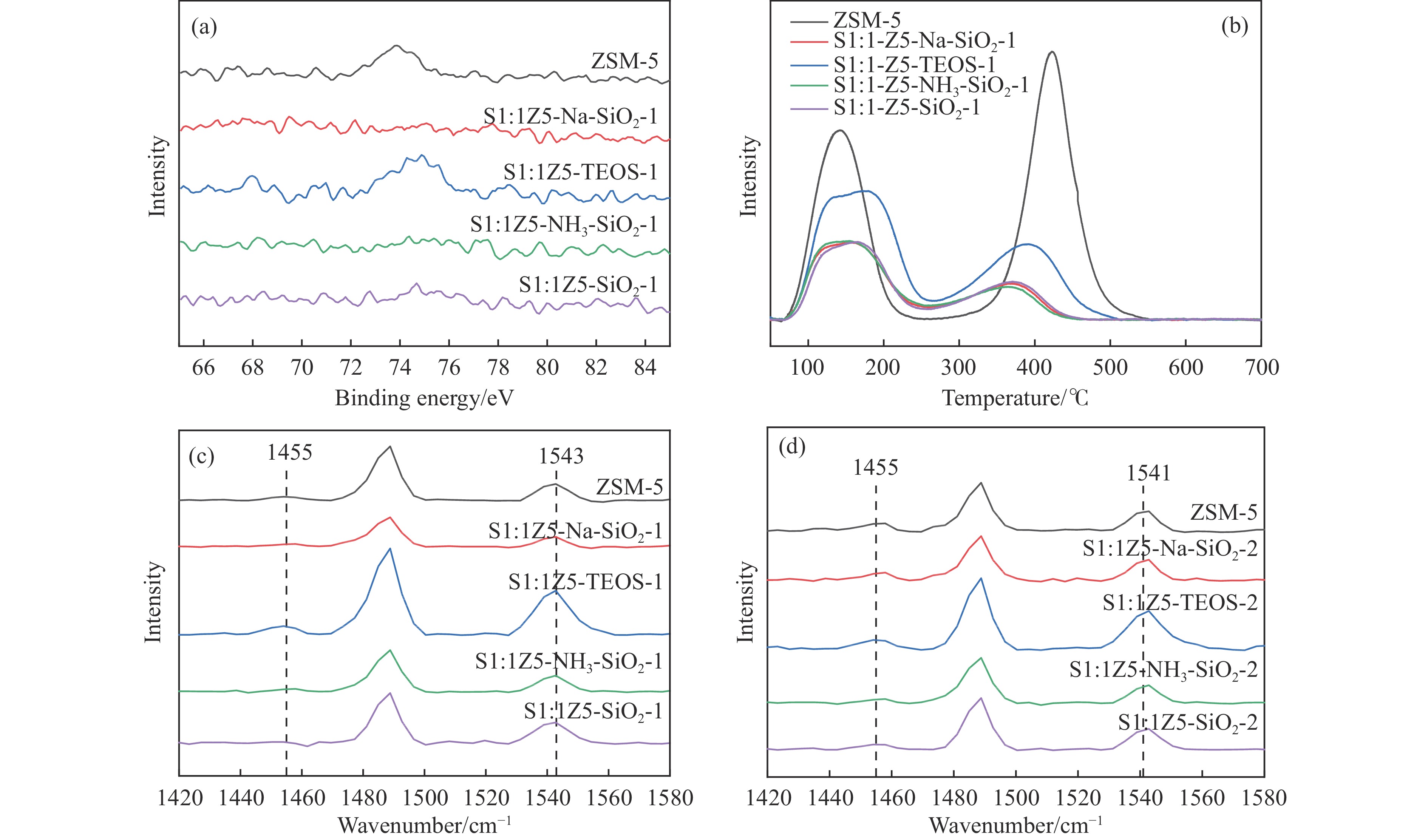
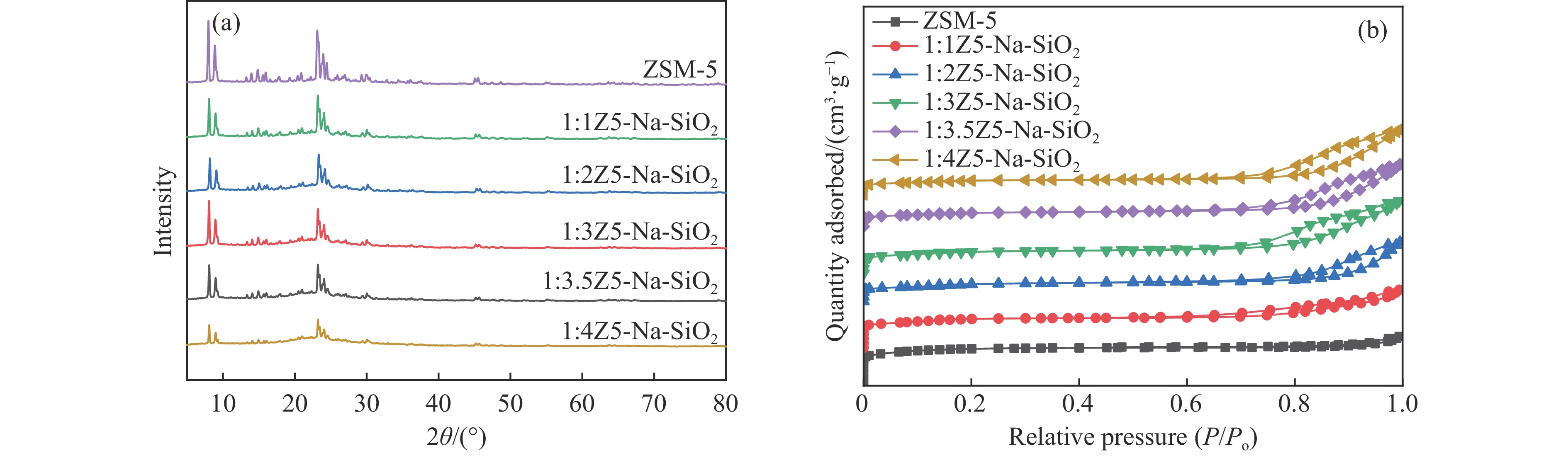
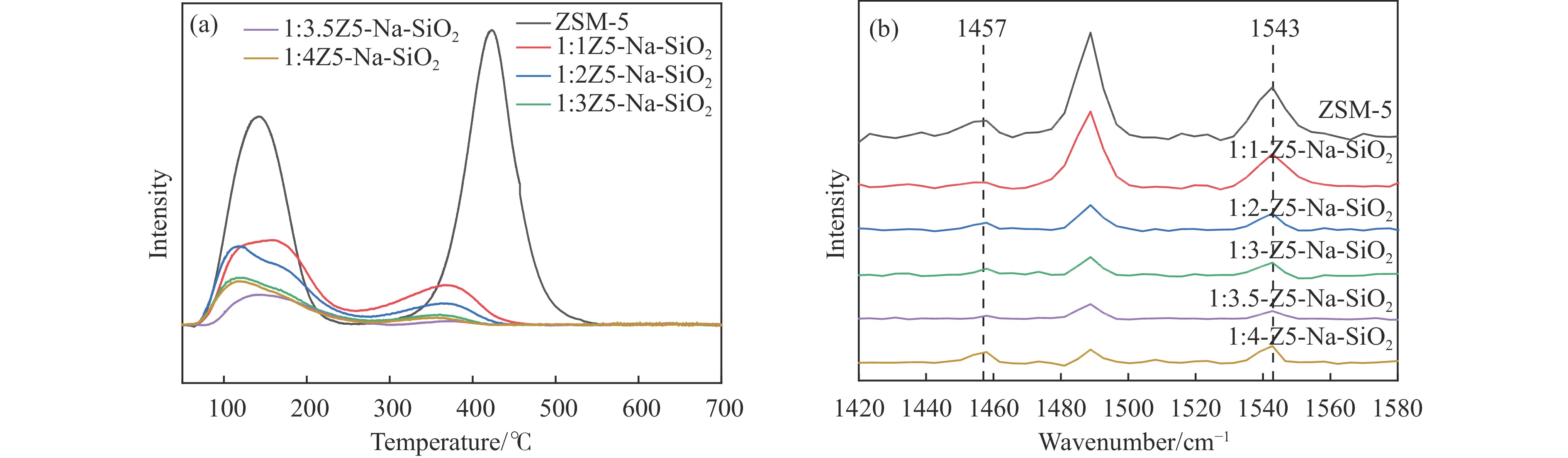
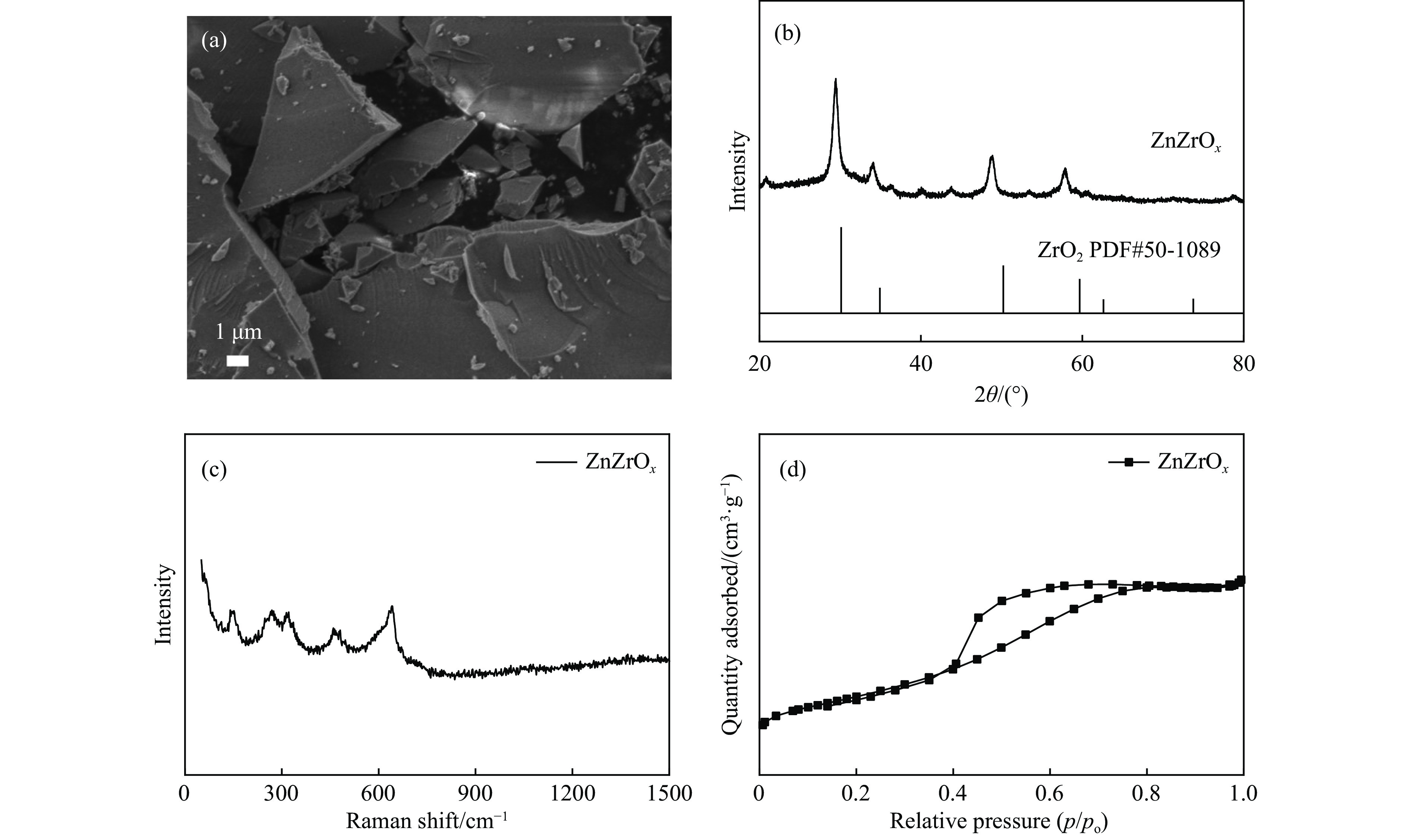
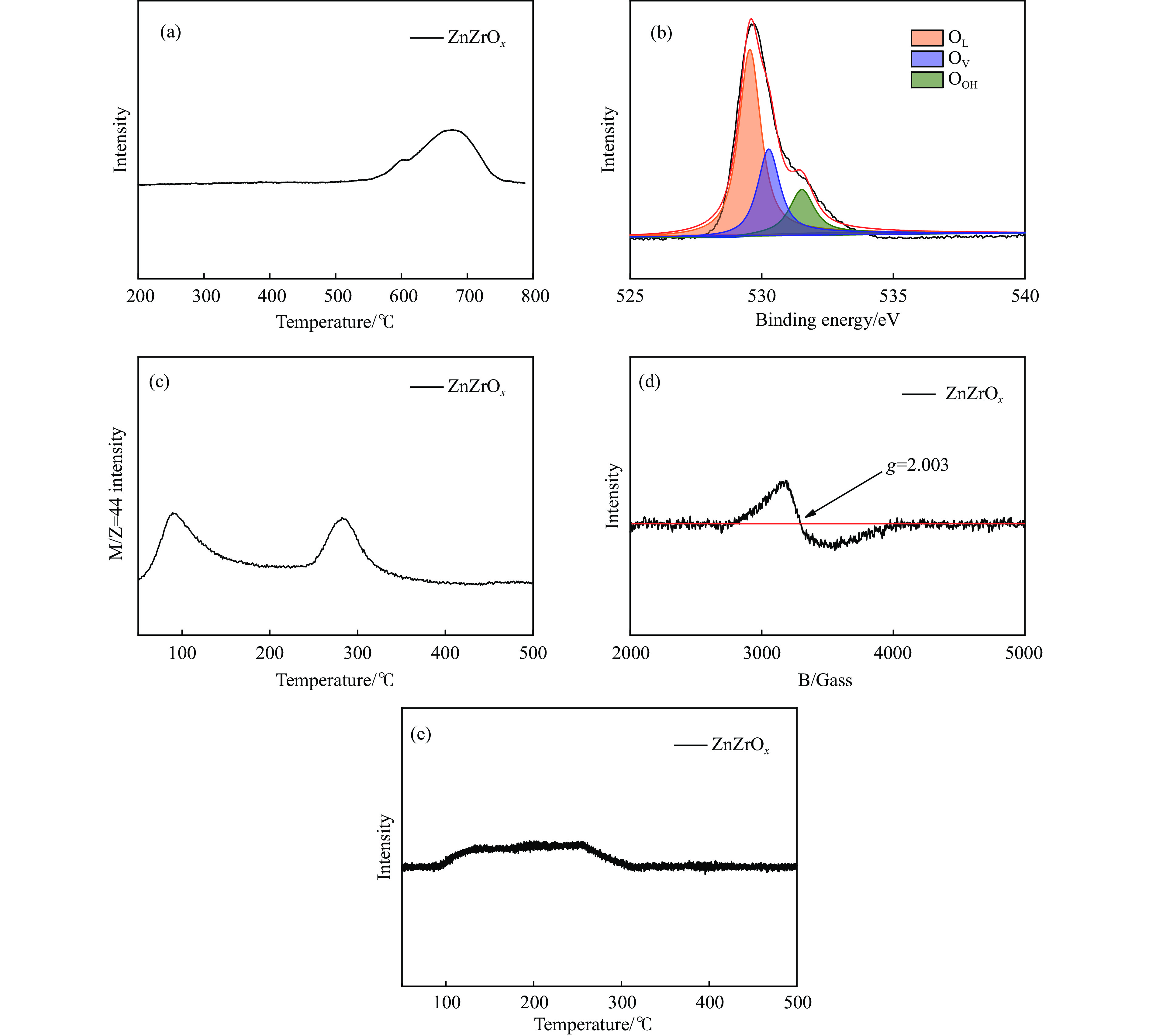
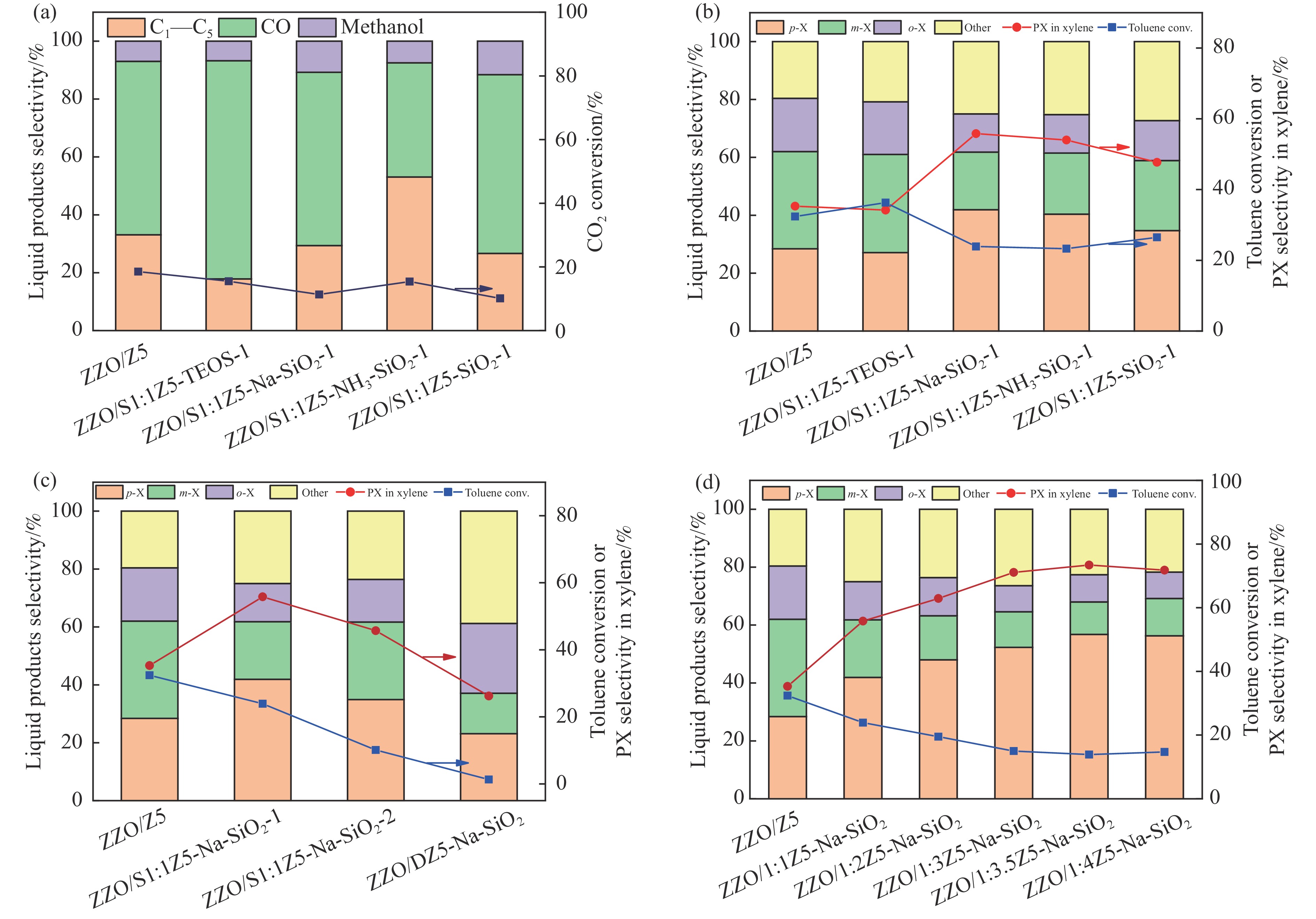
CO2加氢合成高附加值的芳烃对于缓解CO2排放引起的能源气候问题具有重要意义。本研究采用固相法在ZSM-5表面外延生长Silicalite-1,制备出ZSM-5@Silicalite-1分子筛。同时制备高活性氧化物ZnZrOx,并与ZSM-5@Silicalite-1物理混合组成ZnZrOx/ ZSM-5@Silicalite-1双功能催化剂,研究了CO2加氢耦合甲苯烷基化催化性能。相比于ZnZrOx/ZSM-5催化剂,分子筛改性后的双功能催化剂提高了对二甲苯(PX)选择性。研究了晶化条件(硅源、晶化过程、晶化次数)对ZSM-5外延生长Silicalite-1的影响,以及Silicalite-1钝化层厚度对CO2加氢耦合甲苯烷基化反应性能的影响。在400 ℃、3 MPa反应条件下,ZZO/1:3.5Z5-Na-SiO2催化剂的甲苯转化率为12.0%,二甲苯选择性为77.4%,在二甲苯中对二甲苯选择性为73.4%。通过SEM、XRD、N2吸附-脱附、XPS、NH3-TPD、Py-FTIR等表征,研究了分子筛的结构和酸性质。结果表明,通过固相外延生长,延长ZSM-5的孔道,增加间二甲苯(MX)、邻二甲苯(OX)的扩散阻力,同时钝化外表面的酸性,可以有效提高对二甲苯(PX)的选择性。固相外延生长法改性ZSM-5分子筛,摒弃了以往堵塞孔以缩小孔口改性分子筛的缺点,在保证催化剂活性的同时提高了产物选择性。
CO2加氢合成高附加值的芳烃对于缓解CO2排放引起的能源气候问题具有重要意义。本研究采用固相法在ZSM-5表面外延生长Silicalite-1,制备出ZSM-5@Silicalite-1分子筛。同时制备高活性氧化物ZnZrOx,并与ZSM-5@Silicalite-1物理混合组成ZnZrOx/ ZSM-5@Silicalite-1双功能催化剂,研究了CO2加氢耦合甲苯烷基化催化性能。相比于ZnZrOx/ZSM-5催化剂,分子筛改性后的双功能催化剂提高了对二甲苯(PX)选择性。研究了晶化条件(硅源、晶化过程、晶化次数)对ZSM-5外延生长Silicalite-1的影响,以及Silicalite-1钝化层厚度对CO2加氢耦合甲苯烷基化反应性能的影响。在400 ℃、3 MPa反应条件下,ZZO/1:3.5Z5-Na-SiO2催化剂的甲苯转化率为12.0%,二甲苯选择性为77.4%,在二甲苯中对二甲苯选择性为73.4%。通过SEM、XRD、N2吸附-脱附、XPS、NH3-TPD、Py-FTIR等表征,研究了分子筛的结构和酸性质。结果表明,通过固相外延生长,延长ZSM-5的孔道,增加间二甲苯(MX)、邻二甲苯(OX)的扩散阻力,同时钝化外表面的酸性,可以有效提高对二甲苯(PX)的选择性。固相外延生长法改性ZSM-5分子筛,摒弃了以往堵塞孔以缩小孔口改性分子筛的缺点,在保证催化剂活性的同时提高了产物选择性。


当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024004
摘要:
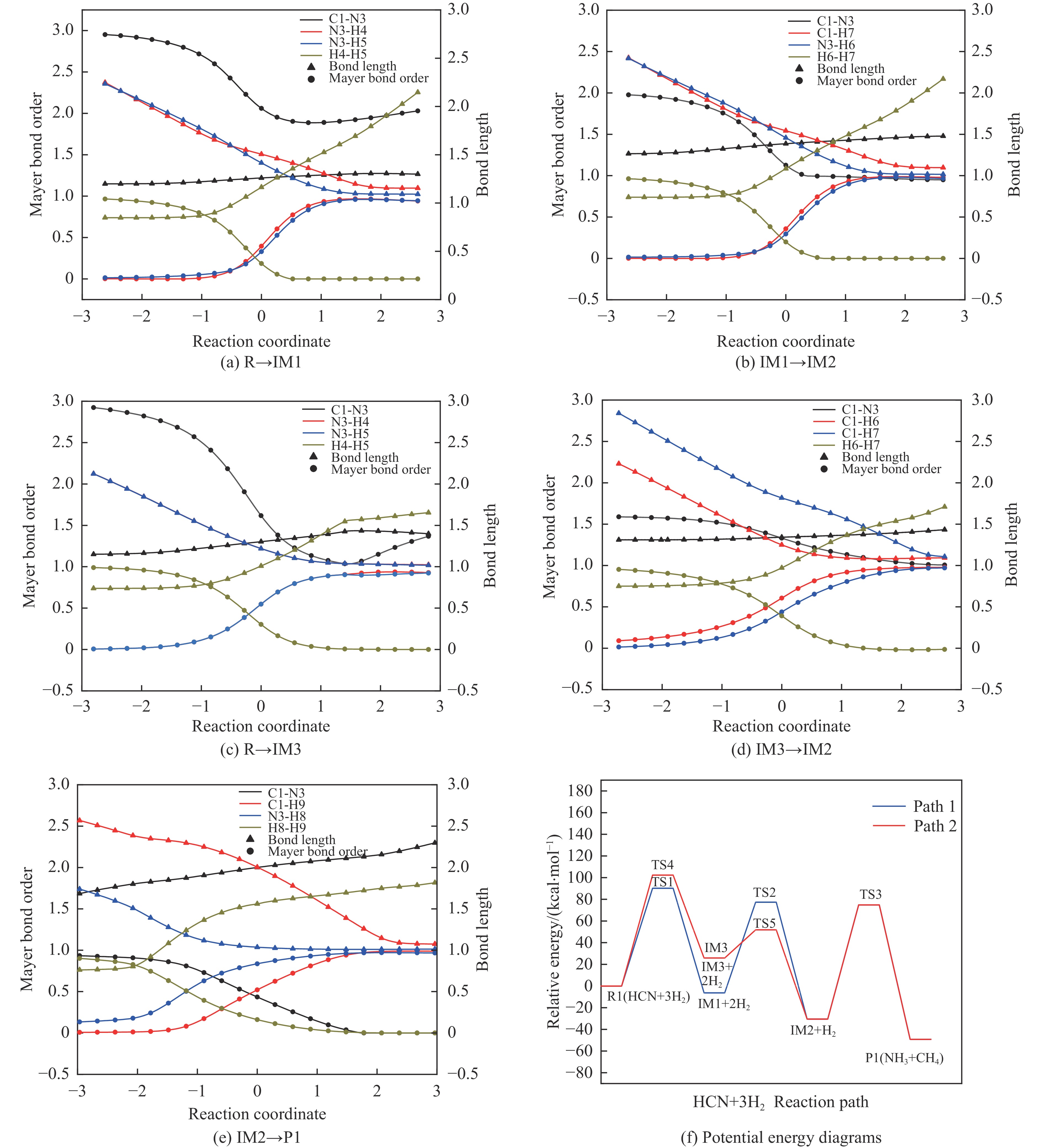
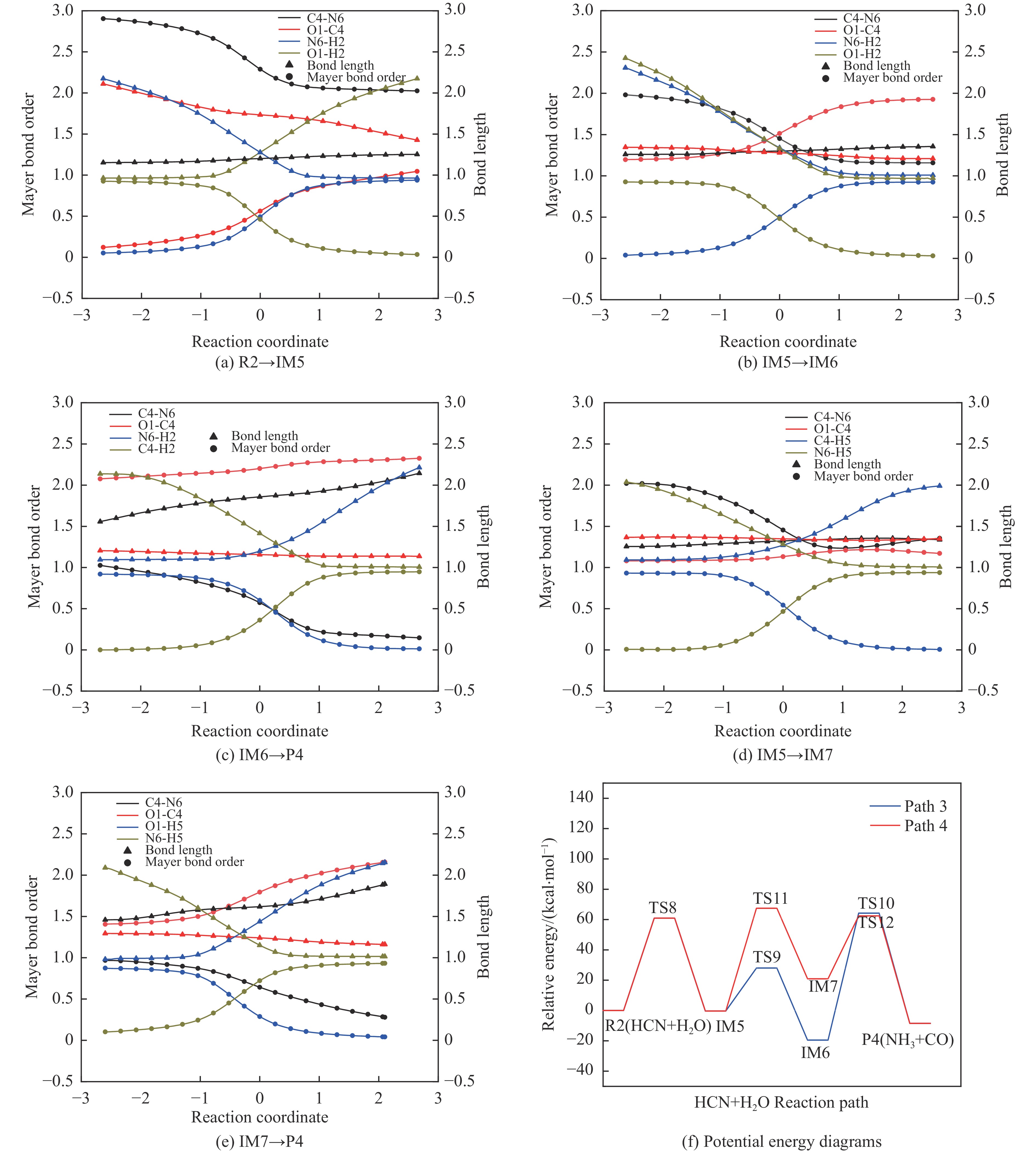
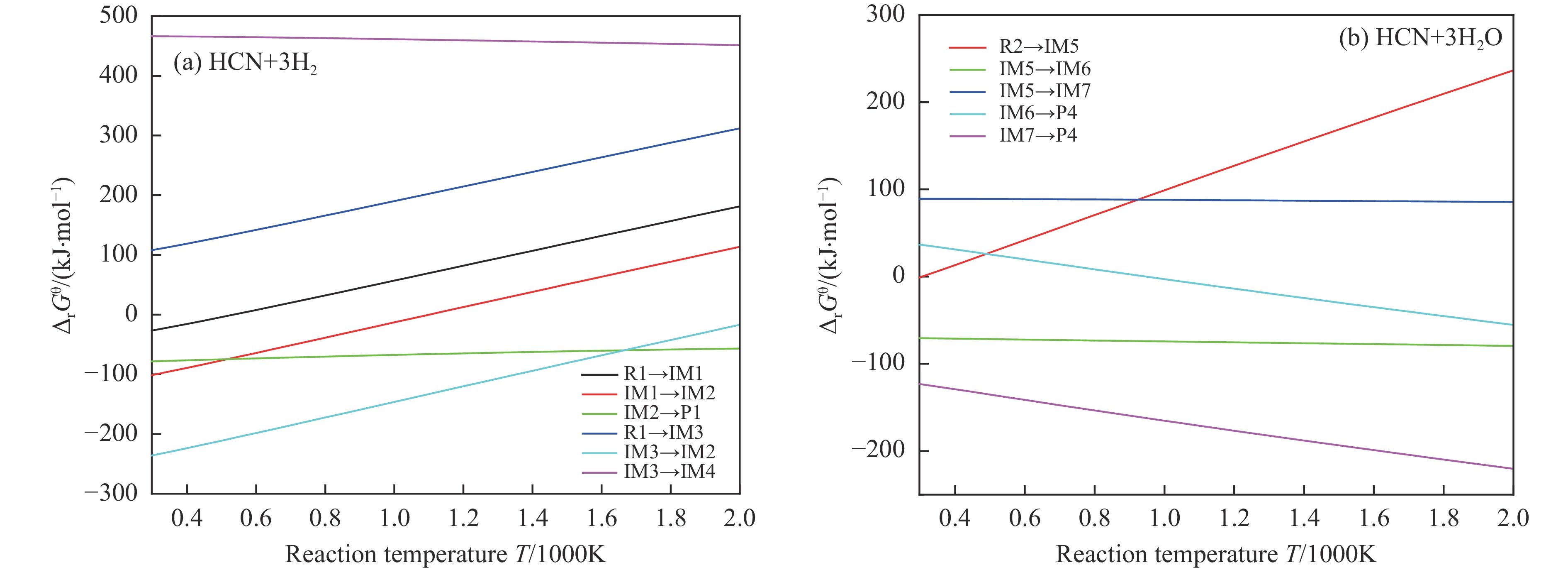
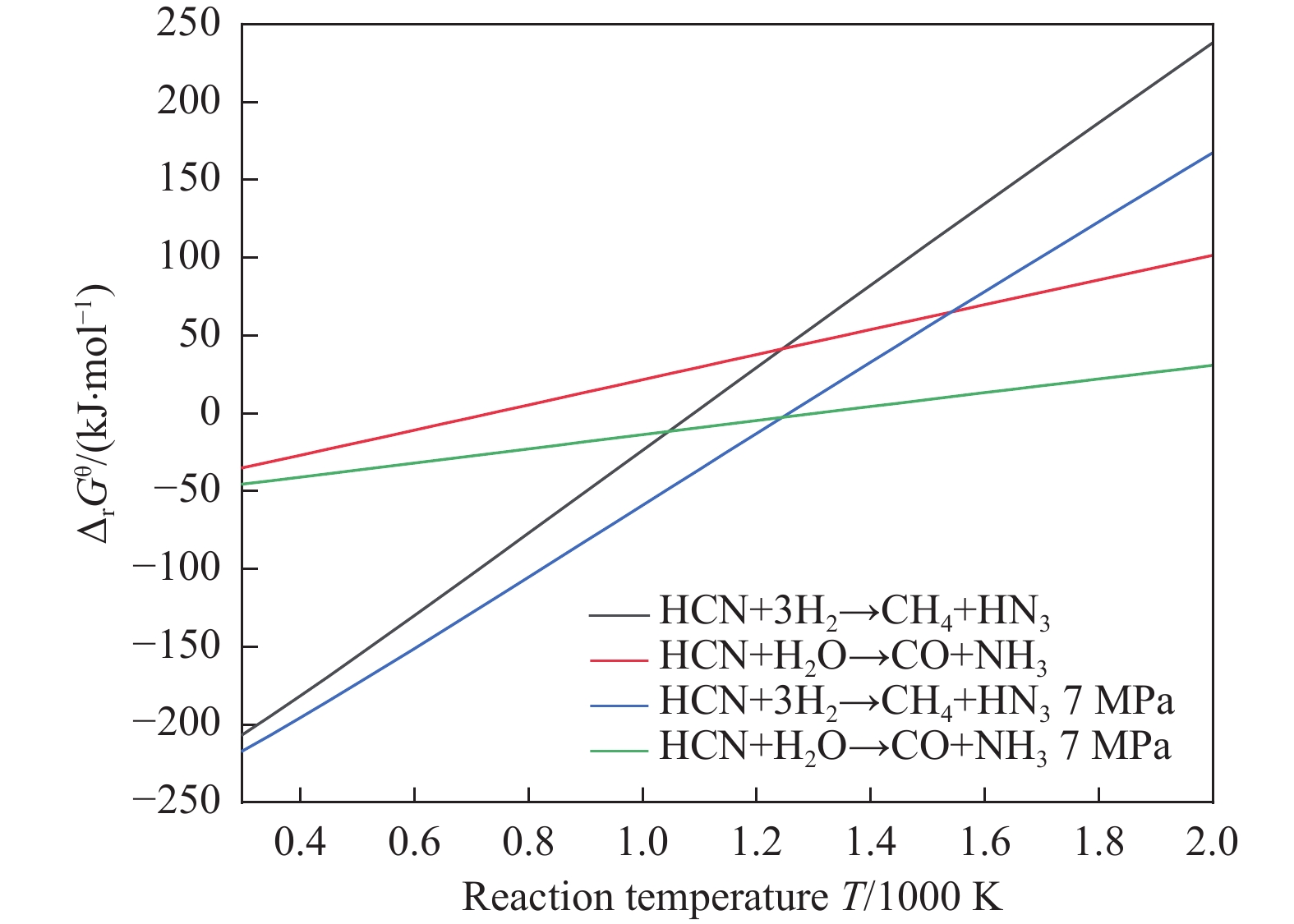
含HCN的废弃物在气化炉内的高温转化是其绿色处理的方法之一,其中,HCN与H2/H2O的反应是其在气化炉内的主要转化过程。本工作基于密度泛函理论,采用Gaussian及其配套软件对HCN与H2/H2O的反应机理进行了研究。通过分子成键、断键角度提出HCN与H2/H2O的各两种反应路径,结合能垒和热力学分析确定了相对最优路径,并计算了相对最优反应路径的速率常数。结果表明,HCN与H2反应相对最优路径为:三个H2分子在C≡N上分三步进行加成得到产物CH4+NH3;HCN与H2O反应相对最优路径为:H2O分子进攻C原子,O原子和C原子的H先后转移至N原子得到产物CO+NH3。两条相对最优路径在1473 K以上有明显反应速率,分别为9.57×10−4和1.71 mol/(L·s)。研究结果为高温下HCN与H2/H2O反应的工艺和设备开发提供了理论数据支撑。
含HCN的废弃物在气化炉内的高温转化是其绿色处理的方法之一,其中,HCN与H2/H2O的反应是其在气化炉内的主要转化过程。本工作基于密度泛函理论,采用Gaussian及其配套软件对HCN与H2/H2O的反应机理进行了研究。通过分子成键、断键角度提出HCN与H2/H2O的各两种反应路径,结合能垒和热力学分析确定了相对最优路径,并计算了相对最优反应路径的速率常数。结果表明,HCN与H2反应相对最优路径为:三个H2分子在C≡N上分三步进行加成得到产物CH4+NH3;HCN与H2O反应相对最优路径为:H2O分子进攻C原子,O原子和C原子的H先后转移至N原子得到产物CO+NH3。两条相对最优路径在1473 K以上有明显反应速率,分别为9.57×10−4和1.71 mol/(L·s)。研究结果为高温下HCN与H2/H2O反应的工艺和设备开发提供了理论数据支撑。

当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60390-1
摘要:
The TiO2 nanotubes arrays/SnO2-Sb (TNTs/SnO2-Sb) electrode is successfully fabricated using the solvothermal synthesis technique. Various architectures of TNTs are constructed by varying the anodization voltage and time, aiming to investigate their impact on the structural and electrochemical properties of the SnO2-Sb electrode. The anodization voltage is identified as the primary influencing factor on the morphology and surface hydrophilia of TNTs arrays, which is evidenced by scanning electron microscopy (SEM) and contact angle testing. In contrast, the effect of anodization time is relatively small. SEM, X-ray diffraction (XRD), linear sweep voltammograms (LSV), and electrochemical impedance spectroscopy (EIS) results indicate that the morphology and crystal size of the catalytic coating, as well as the oxygen evolution potential of the electrode, are influenced by the pore size of TNTs arrays. The influencing mechanism of enhanced electrochemical activity by adjusting the architecture of TNTs arrays is investigated using X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR), and hydroxyl radicals (·OH) generation test. The results reveal a higher concentration of oxygen vacancies on the sample with a compact and smaller particle coating, indicating the presence of more adsorbed oxygen species. Consequently, this enhances the generation capacity of active radicals for organic matter degradation. The electrode featuring TNTs arrays with a length of 950 nm and a pore diameter of 100 nm exhibits the most effective remediation of phenol-containing wastewater, achieving approximately 92% ± 4.6% removal after a duration of 2 h.
The TiO2 nanotubes arrays/SnO2-Sb (TNTs/SnO2-Sb) electrode is successfully fabricated using the solvothermal synthesis technique. Various architectures of TNTs are constructed by varying the anodization voltage and time, aiming to investigate their impact on the structural and electrochemical properties of the SnO2-Sb electrode. The anodization voltage is identified as the primary influencing factor on the morphology and surface hydrophilia of TNTs arrays, which is evidenced by scanning electron microscopy (SEM) and contact angle testing. In contrast, the effect of anodization time is relatively small. SEM, X-ray diffraction (XRD), linear sweep voltammograms (LSV), and electrochemical impedance spectroscopy (EIS) results indicate that the morphology and crystal size of the catalytic coating, as well as the oxygen evolution potential of the electrode, are influenced by the pore size of TNTs arrays. The influencing mechanism of enhanced electrochemical activity by adjusting the architecture of TNTs arrays is investigated using X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR), and hydroxyl radicals (·OH) generation test. The results reveal a higher concentration of oxygen vacancies on the sample with a compact and smaller particle coating, indicating the presence of more adsorbed oxygen species. Consequently, this enhances the generation capacity of active radicals for organic matter degradation. The electrode featuring TNTs arrays with a length of 950 nm and a pore diameter of 100 nm exhibits the most effective remediation of phenol-containing wastewater, achieving approximately 92% ± 4.6% removal after a duration of 2 h.












当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023084
摘要:
选择性催化还原技术(SCR)在水泥行业脱硝中被广泛应用,其中,高温范围内(280–350 ℃)已有较为完善的SCR技术及体系,但在中温区仍有待突破。本工作以中温脱硝催化剂为重点,综述了Mn、Ce、V系脱硝催化剂的研究进展,并分析了Sm、Nb、Ho、Sb、La、Mo、Pr的掺杂对于脱硝催化剂的改性,结合水泥窑炉烟尘中SO2、H2O、碱金属含量高的特点,分析了脱硝催化剂中毒原因,对催化剂的抗H2O、SO2、碱金属中毒性能进行了探讨,展望了水泥行业SCR中温脱硝催化剂的研究前景。
选择性催化还原技术(SCR)在水泥行业脱硝中被广泛应用,其中,高温范围内(280–350 ℃)已有较为完善的SCR技术及体系,但在中温区仍有待突破。本工作以中温脱硝催化剂为重点,综述了Mn、Ce、V系脱硝催化剂的研究进展,并分析了Sm、Nb、Ho、Sb、La、Mo、Pr的掺杂对于脱硝催化剂的改性,结合水泥窑炉烟尘中SO2、H2O、碱金属含量高的特点,分析了脱硝催化剂中毒原因,对催化剂的抗H2O、SO2、碱金属中毒性能进行了探讨,展望了水泥行业SCR中温脱硝催化剂的研究前景。









当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60403-7
摘要:
生物质基2,5-呋喃二甲醇(BHMF)可从廉价易得的糖类出发,经催化转化-选择性氢化制取,并作为一种用途广泛的化工中间体及燃料前体,尤其在改善传统聚酯性能以及合成绿色可降解的生物基聚酯新材料方面具有独特优势。BHMF制取过程中,传统的氢化方式消耗了大量高品位能源氢气,且高压氢气存在安全隐患并导致基础设施投入多。本工作立足于催化转移氢化的优势,综述了甲酸、醇类及其他类型氢供体通过催化转移氢化的方式选择性加氢制取BHMF的研究进展;并针对催化转移氢化过程中不同类型氢供体、催化剂和反应工艺的特点及存在的问题,分析了反应条件、强化手段等对BHMF选择性和收率的影响以及反应体系的优劣。在此基础上,提出了转移氢化制取BHMF新型催化体系的研究方向,并对清洁高效、本质安全BHMF制取工艺的发展进行了展望,为生物质转化中特定催化体系的研发提供科学参考。
生物质基2,5-呋喃二甲醇(BHMF)可从廉价易得的糖类出发,经催化转化-选择性氢化制取,并作为一种用途广泛的化工中间体及燃料前体,尤其在改善传统聚酯性能以及合成绿色可降解的生物基聚酯新材料方面具有独特优势。BHMF制取过程中,传统的氢化方式消耗了大量高品位能源氢气,且高压氢气存在安全隐患并导致基础设施投入多。本工作立足于催化转移氢化的优势,综述了甲酸、醇类及其他类型氢供体通过催化转移氢化的方式选择性加氢制取BHMF的研究进展;并针对催化转移氢化过程中不同类型氢供体、催化剂和反应工艺的特点及存在的问题,分析了反应条件、强化手段等对BHMF选择性和收率的影响以及反应体系的优劣。在此基础上,提出了转移氢化制取BHMF新型催化体系的研究方向,并对清洁高效、本质安全BHMF制取工艺的发展进行了展望,为生物质转化中特定催化体系的研发提供科学参考。




















当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023087
摘要:
以Co基催化剂耦合沸石分子筛催化剂应用于合成气催化转化可以有效改善催化剂的产物选择性。本研究通过浸渍法制备得到Zr/Al2O3载体和Pt/ZSM-5催化剂,再通过超声分散法制备了Co/Al2O3、Co/Zr/Al2O3和Co/Zr/Al2O3-Pt/ZSM-5催化剂。通过系列表征技术对载体和催化剂理化性质进行分析,评价了催化剂费-托合成反应性能。结果表明,Zr的引入有助于提升Co/Zr/Al2O3上Co物种的还原性,改善催化活性,增加C12+重质烃的选择性。当Co/Zr/Al2O3与Pt/ZSM-5耦合后,由于贵金属Pt的助剂效应,进一步促进Co物种的还原,Co/Zr/Al2O3-Pt/ZSM-5催化剂的CTY值提高至8.3×10−5 mmol/(g·s),同时具有较低的CH4、C2−C4产物选择性。此外,Pt/ZSM-5的酸性促进C12+产物的部分裂解,使产物分布向C5−C11液态烃偏移,C5−C11产物选择性达到45.9%。本研究为设计和制备高效的费-托合成催化剂提供了参考。
以Co基催化剂耦合沸石分子筛催化剂应用于合成气催化转化可以有效改善催化剂的产物选择性。本研究通过浸渍法制备得到Zr/Al2O3载体和Pt/ZSM-5催化剂,再通过超声分散法制备了Co/Al2O3、Co/Zr/Al2O3和Co/Zr/Al2O3-Pt/ZSM-5催化剂。通过系列表征技术对载体和催化剂理化性质进行分析,评价了催化剂费-托合成反应性能。结果表明,Zr的引入有助于提升Co/Zr/Al2O3上Co物种的还原性,改善催化活性,增加C12+重质烃的选择性。当Co/Zr/Al2O3与Pt/ZSM-5耦合后,由于贵金属Pt的助剂效应,进一步促进Co物种的还原,Co/Zr/Al2O3-Pt/ZSM-5催化剂的CTY值提高至8.3×10−5 mmol/(g·s),同时具有较低的CH4、C2−C4产物选择性。此外,Pt/ZSM-5的酸性促进C12+产物的部分裂解,使产物分布向C5−C11液态烃偏移,C5−C11产物选择性达到45.9%。本研究为设计和制备高效的费-托合成催化剂提供了参考。











当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023085
摘要:
通过简单加热乙二醇,对甲苯磺酸和八水氯氧化锆混合物制备了三元低共熔溶剂。采用傅里叶变换红外光谱(FT-IR)和核磁共振氢谱(1H NMR)验证了低共熔溶剂成功合成。分别采用紫外-可见吸收光谱和旋转式黏度计对其酸性和黏度进行测试。以双氧水作为氧化剂,以合成的低共熔溶剂为萃取剂和催化剂构成萃取-氧化脱硫系统,考察了低共熔溶剂的组成、反应温度、氧硫比、剂油比以及不同硫化物等对脱硫率的影响。实验结果表明,在氯氧化锆、乙二醇和对苯甲磺酸物质的量比为1∶10∶10,反应温度50 ℃、剂油比为1∶5、氧硫比为8的最佳反应条件下,二苯并噻吩(DBT)、4,6-二甲基二苯并噻吩(4,6-DMDBT)、苯并噻吩(BT)模拟油的脱硫率分别为100%、92.2%、60%,且低共熔溶剂重复使用五次后脱硫率仍可达到96.2%,最后对氧化脱硫的机理进行了探讨。
通过简单加热乙二醇,对甲苯磺酸和八水氯氧化锆混合物制备了三元低共熔溶剂。采用傅里叶变换红外光谱(FT-IR)和核磁共振氢谱(1H NMR)验证了低共熔溶剂成功合成。分别采用紫外-可见吸收光谱和旋转式黏度计对其酸性和黏度进行测试。以双氧水作为氧化剂,以合成的低共熔溶剂为萃取剂和催化剂构成萃取-氧化脱硫系统,考察了低共熔溶剂的组成、反应温度、氧硫比、剂油比以及不同硫化物等对脱硫率的影响。实验结果表明,在氯氧化锆、乙二醇和对苯甲磺酸物质的量比为1∶10∶10,反应温度50 ℃、剂油比为1∶5、氧硫比为8的最佳反应条件下,二苯并噻吩(DBT)、4,6-二甲基二苯并噻吩(4,6-DMDBT)、苯并噻吩(BT)模拟油的脱硫率分别为100%、92.2%、60%,且低共熔溶剂重复使用五次后脱硫率仍可达到96.2%,最后对氧化脱硫的机理进行了探讨。
















当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60402-5
摘要:
煤气化细渣是煤炭清洁高效利用的副产物之一,其资源化应用迫在眉睫。通过简单筛分得到固定碳含量高于60%的高炭组分,并以此为原料,采用超声酸浸法制备多孔材料。以核废水中放射性碘的吸附处理为应用背景,用碘吸附值表征多孔材料的吸附性能。结合SEM、BET、XRD和FT-IR等性质和结构分析方法,系统研究了超声时间、超声功率、酸浓度和温度对多孔材料碘吸附性能和组成结构的影响规律;并探讨了超声强化酸浸对残炭的组成结构的影响机制和灰成分的迁移转化规律,总结出超声强化酸浸作用机理。结果表明,煤气化细渣高炭组分在酸浓度为4 mol/L、酸浸温度为50 ℃、超声功率为210 W,超声时间1.5 h的条件下,所制备多孔材料的碘吸附性能最佳,为468.53 mg/g,比表面积达到474.97 m2/g,且具有以介孔为主的丰富孔隙结构。各因素对多孔材料碘吸附性能影响的顺序为:超声时间>酸浓度>超声功率>酸浸温度。超声强化酸浸作用机理是超声空化和机械波作用一方面强化炭灰黏附颗粒的解离,使堵塞在气化细渣孔道内的灰颗粒脱附,增加孔隙结构的连通性;其次,会导致炭灰颗粒表面裂纹的产生,增强碳颗粒内部无机组分的可及性;第三,能够提高酸浸过程的传质速率,强化气化细渣中的无机组分的浸出效果。
煤气化细渣是煤炭清洁高效利用的副产物之一,其资源化应用迫在眉睫。通过简单筛分得到固定碳含量高于60%的高炭组分,并以此为原料,采用超声酸浸法制备多孔材料。以核废水中放射性碘的吸附处理为应用背景,用碘吸附值表征多孔材料的吸附性能。结合SEM、BET、XRD和FT-IR等性质和结构分析方法,系统研究了超声时间、超声功率、酸浓度和温度对多孔材料碘吸附性能和组成结构的影响规律;并探讨了超声强化酸浸对残炭的组成结构的影响机制和灰成分的迁移转化规律,总结出超声强化酸浸作用机理。结果表明,煤气化细渣高炭组分在酸浓度为4 mol/L、酸浸温度为50 ℃、超声功率为210 W,超声时间1.5 h的条件下,所制备多孔材料的碘吸附性能最佳,为468.53 mg/g,比表面积达到474.97 m2/g,且具有以介孔为主的丰富孔隙结构。各因素对多孔材料碘吸附性能影响的顺序为:超声时间>酸浓度>超声功率>酸浸温度。超声强化酸浸作用机理是超声空化和机械波作用一方面强化炭灰黏附颗粒的解离,使堵塞在气化细渣孔道内的灰颗粒脱附,增加孔隙结构的连通性;其次,会导致炭灰颗粒表面裂纹的产生,增强碳颗粒内部无机组分的可及性;第三,能够提高酸浸过程的传质速率,强化气化细渣中的无机组分的浸出效果。
















当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60444-5
摘要:
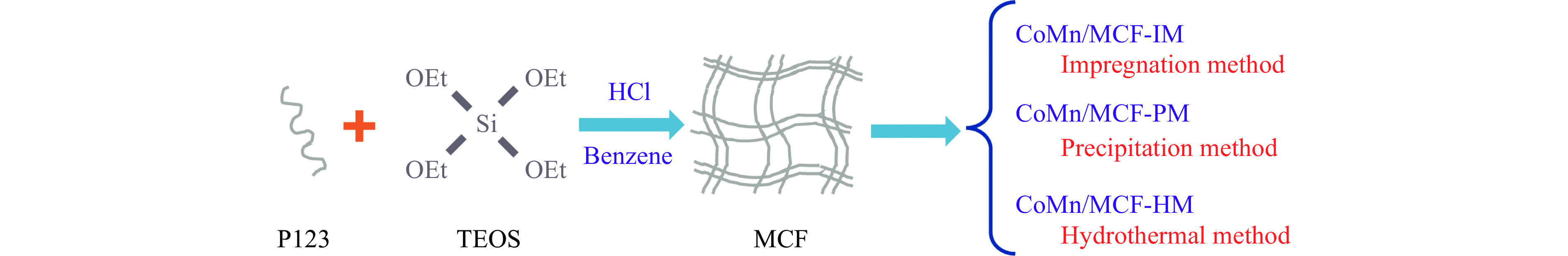
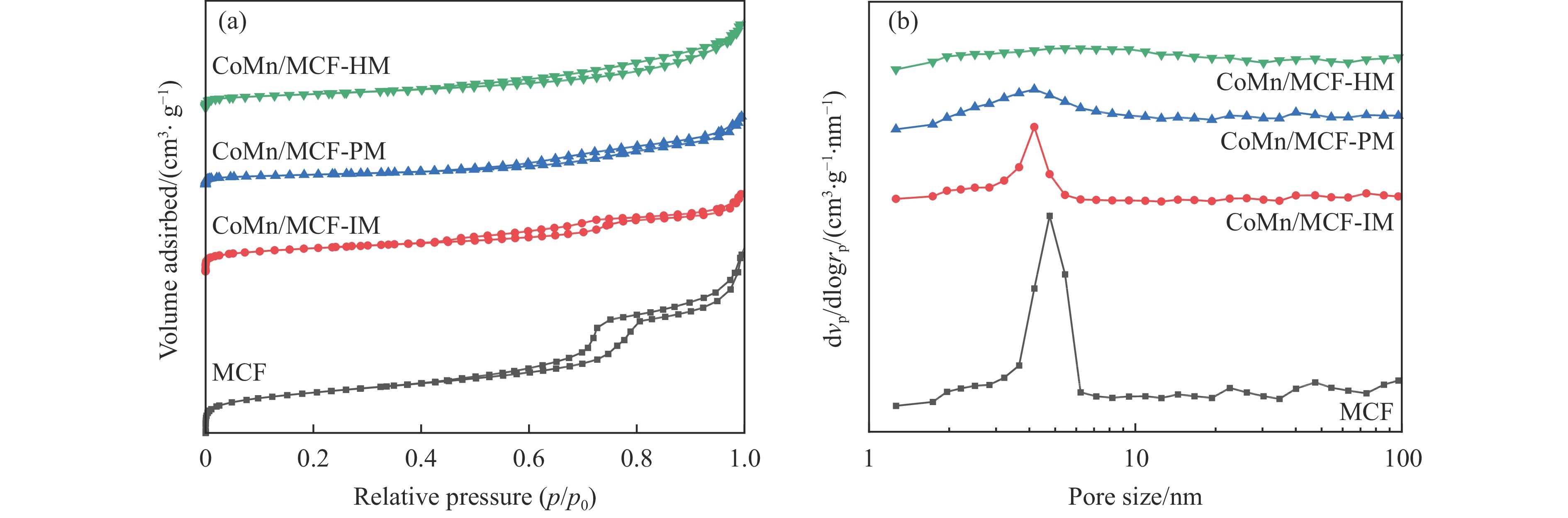
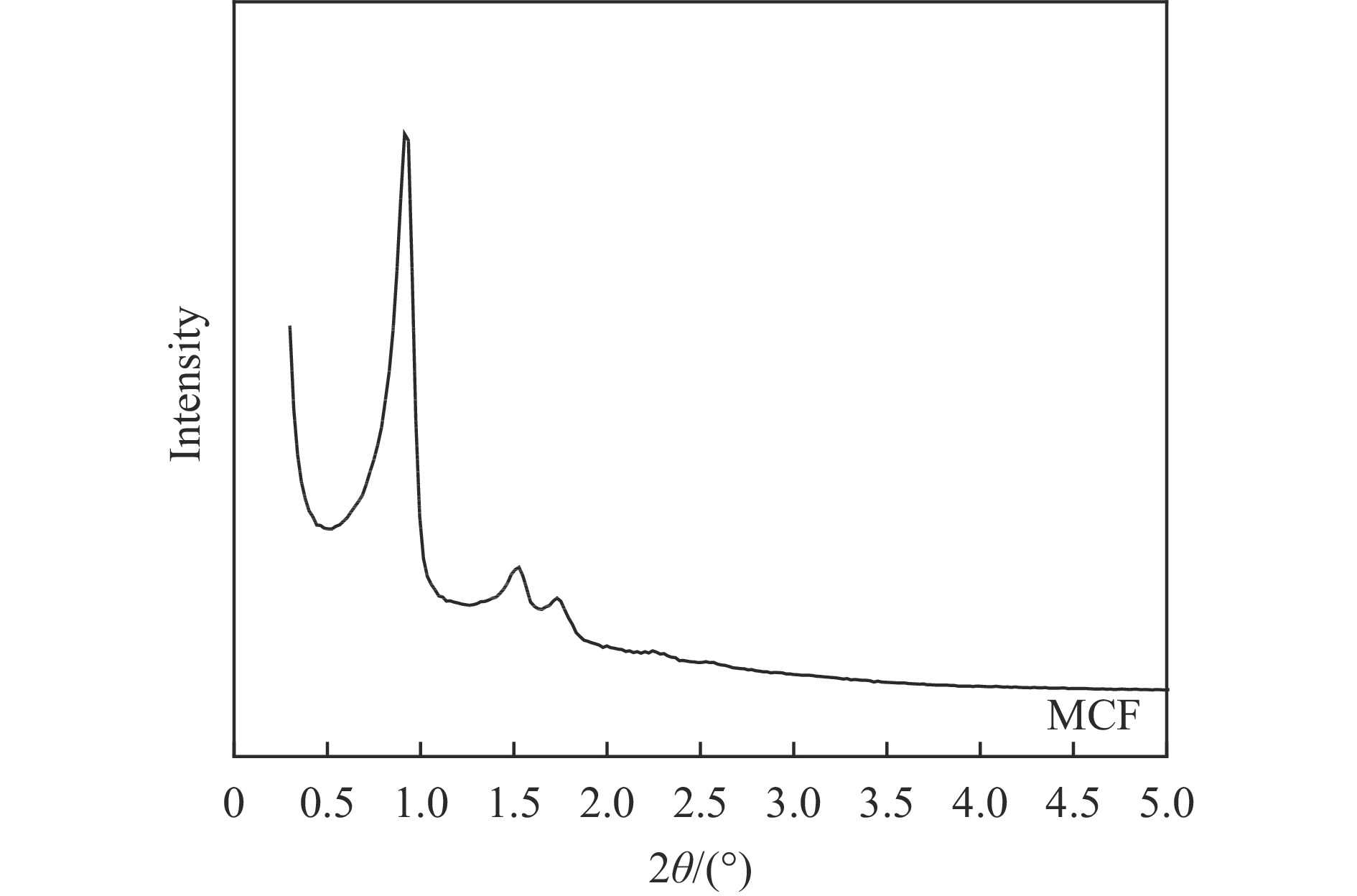
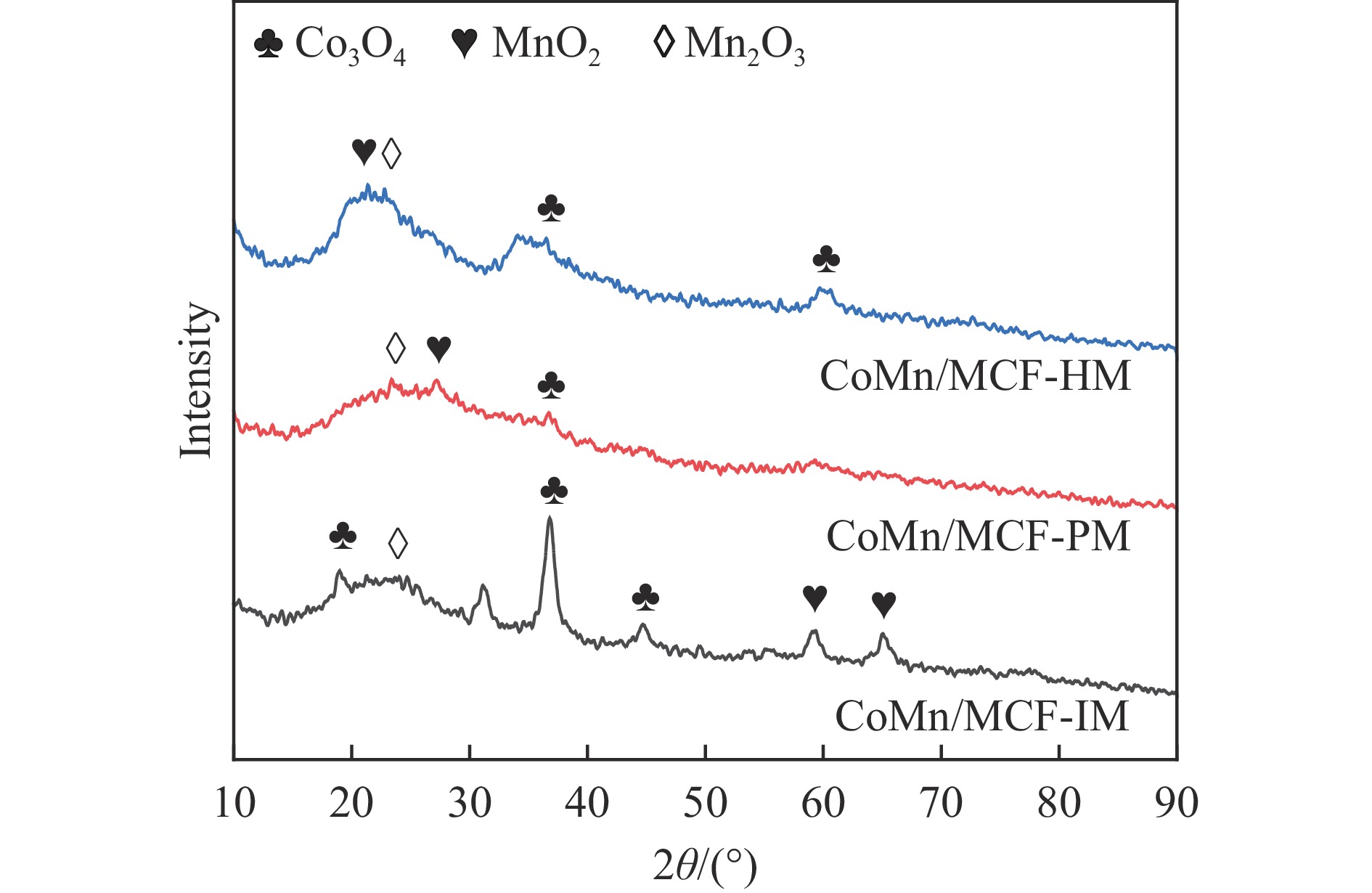
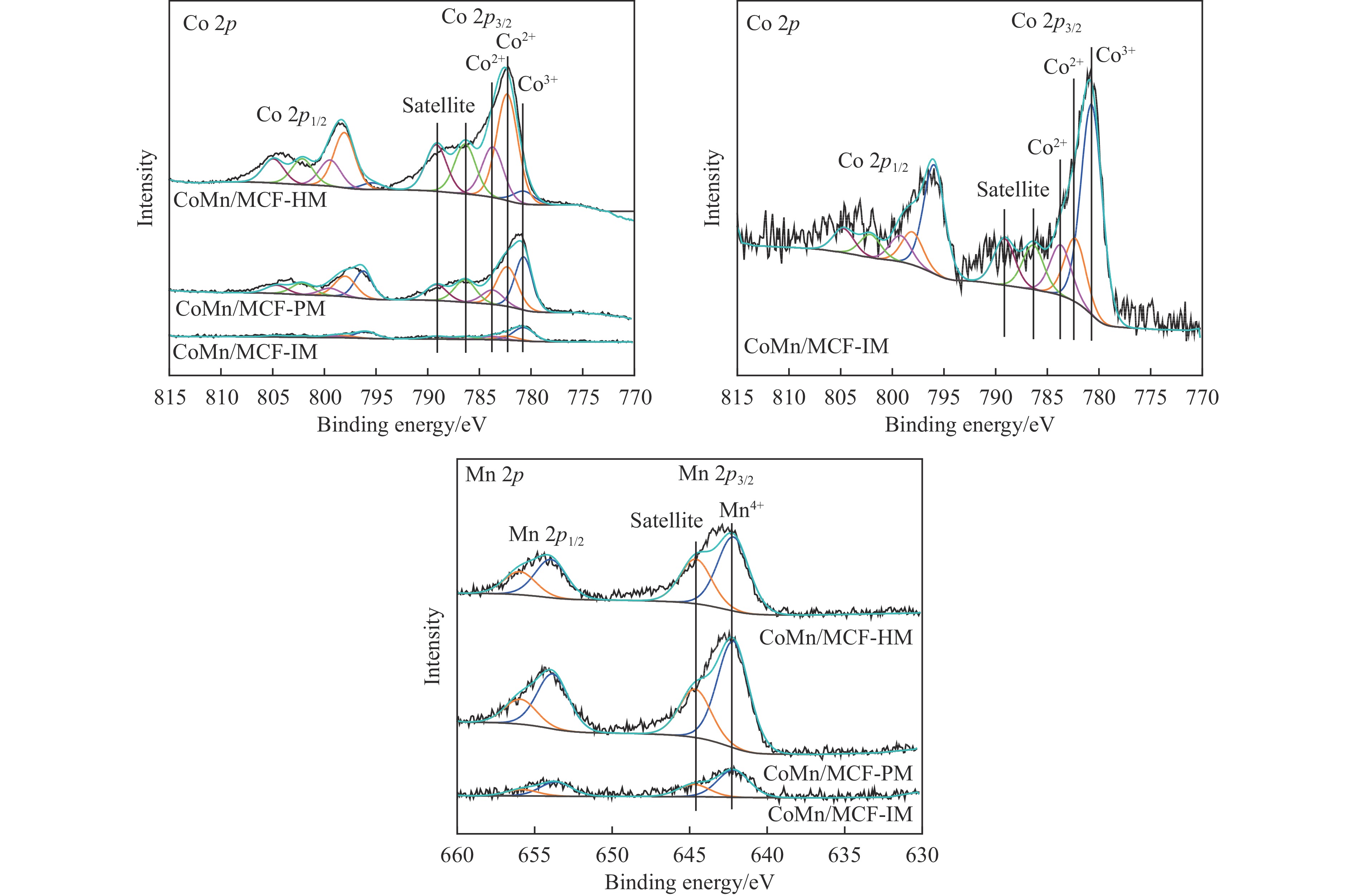
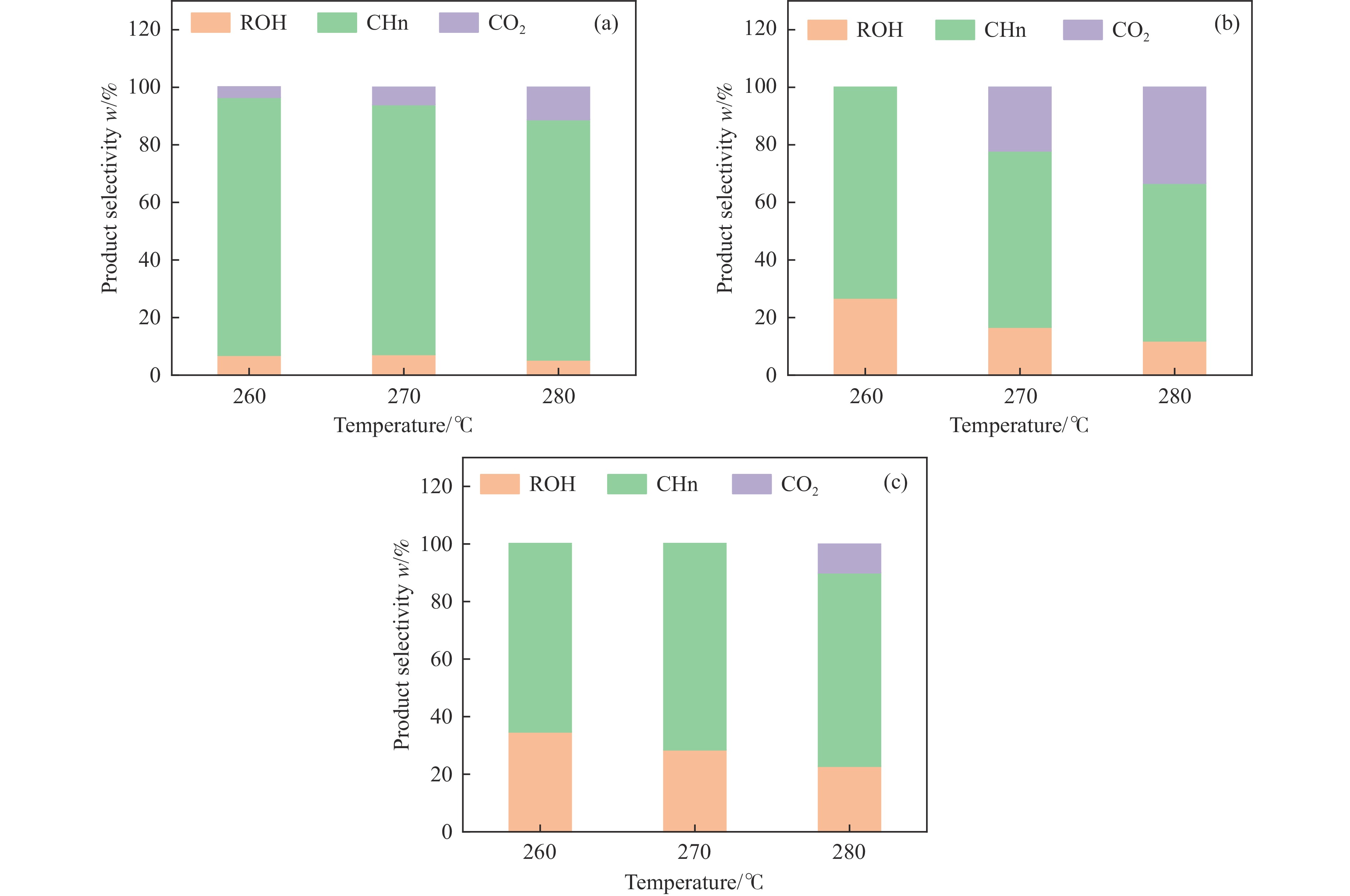
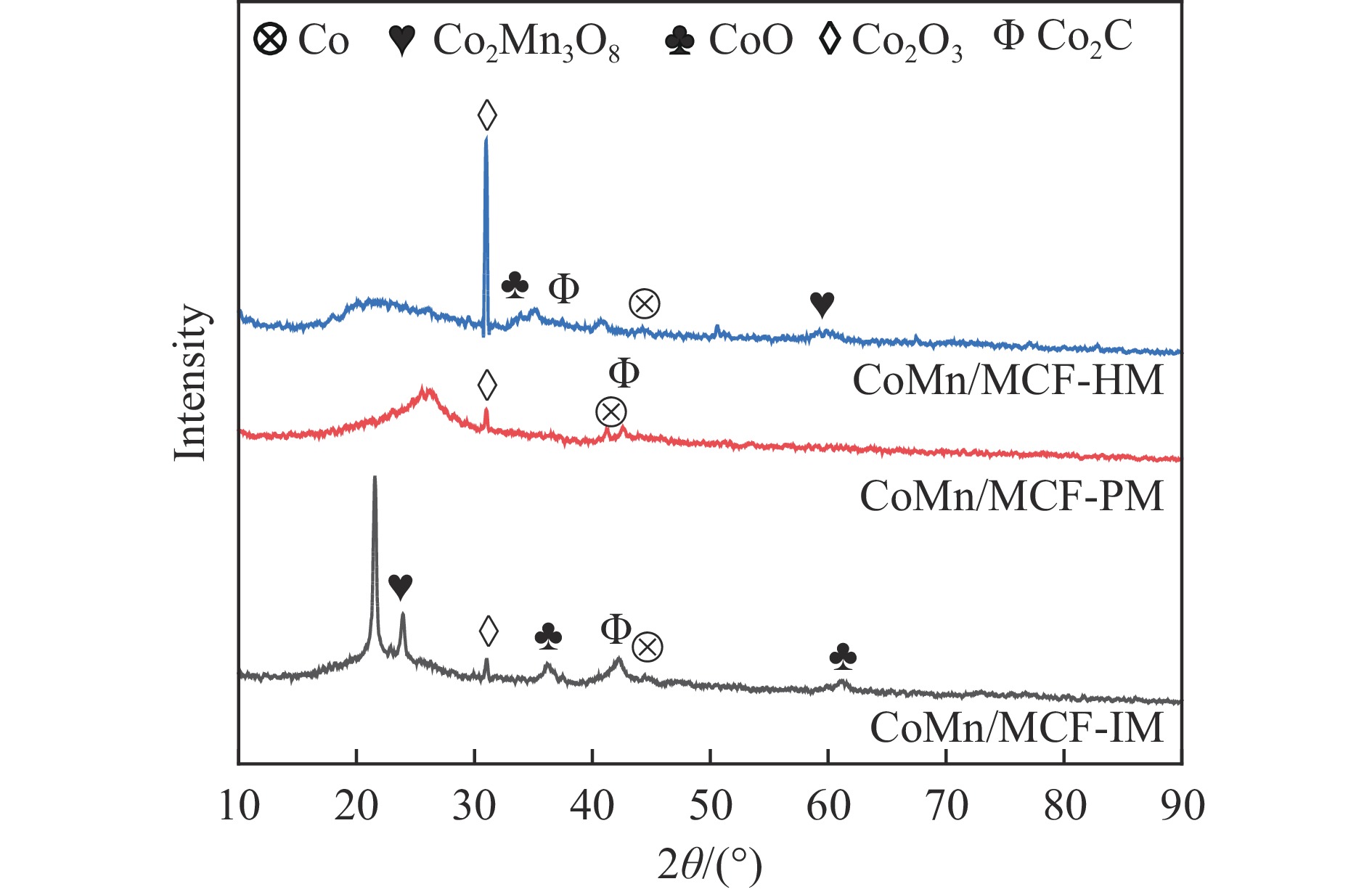
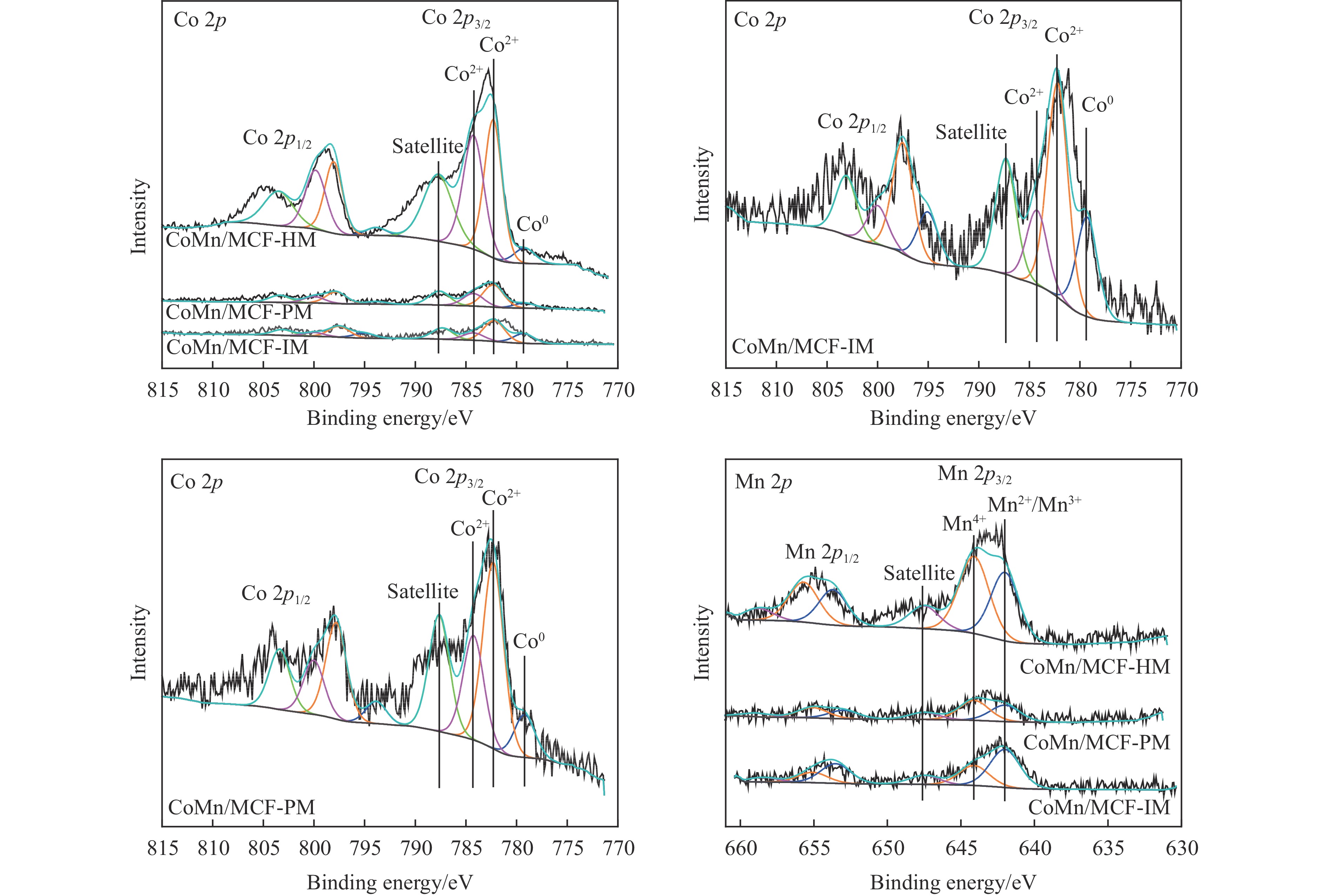
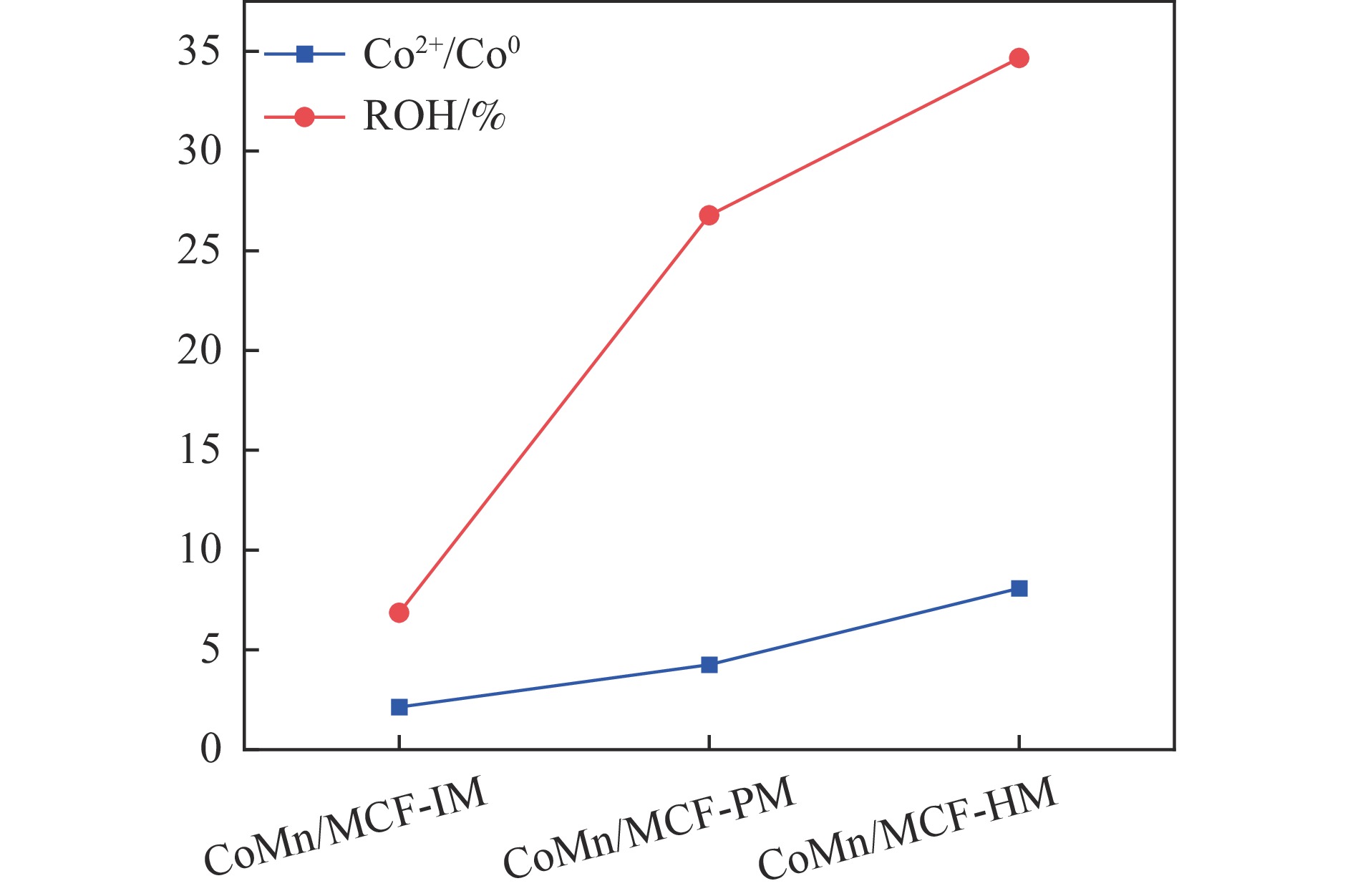
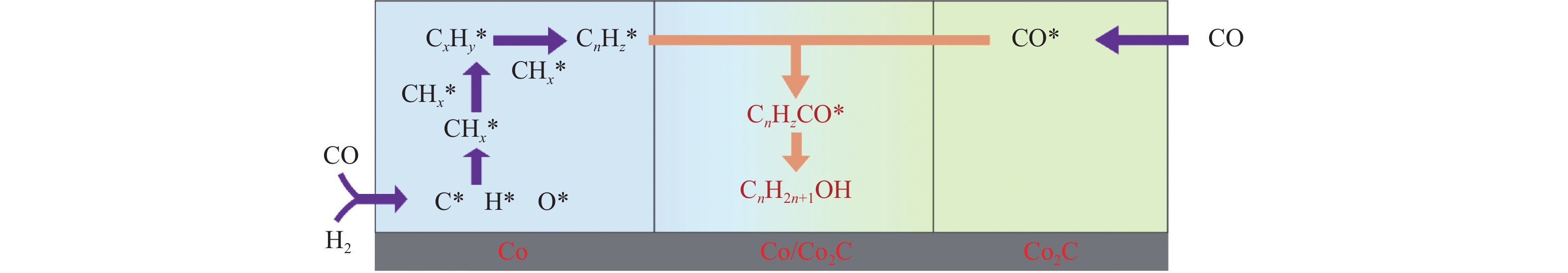
本研究采用浸渍法、沉淀法和水热合成法制备了一系列泡沫硅负载CoMn基催化剂,并结合XRD、H2-TPR、N2物理吸附、TEM和XPS等表征技术考察了制备方法对催化剂在合成气制低碳醇反应中的性能影响。研究表明,催化剂表面存在Co2+(Co2C)、Co0物种,水热合成法制备的催化剂表面Co2C-Co0活性位点存在良好的协同作用有利于醇的生成,较高比例的Co2C也促进了CO的非解离吸附和插入,从而呈现最高的醇选择性。在t=260 ℃,p=5.0 MPa,GHSV=4500 h−1,H2/CO(体积比)=2∶1的反应条件下,该泡沫催化剂可实现CO转化率11.1%,总醇选择性34.7%,C2+OH选择性34.5%的反应性能。
本研究采用浸渍法、沉淀法和水热合成法制备了一系列泡沫硅负载CoMn基催化剂,并结合XRD、H2-TPR、N2物理吸附、TEM和XPS等表征技术考察了制备方法对催化剂在合成气制低碳醇反应中的性能影响。研究表明,催化剂表面存在Co2+(Co2C)、Co0物种,水热合成法制备的催化剂表面Co2C-Co0活性位点存在良好的协同作用有利于醇的生成,较高比例的Co2C也促进了CO的非解离吸附和插入,从而呈现最高的醇选择性。在t=260 ℃,p=5.0 MPa,GHSV=4500 h−1,H2/CO(体积比)=2∶1的反应条件下,该泡沫催化剂可实现CO转化率11.1%,总醇选择性34.7%,C2+OH选择性34.5%的反应性能。


当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60435-4
摘要:
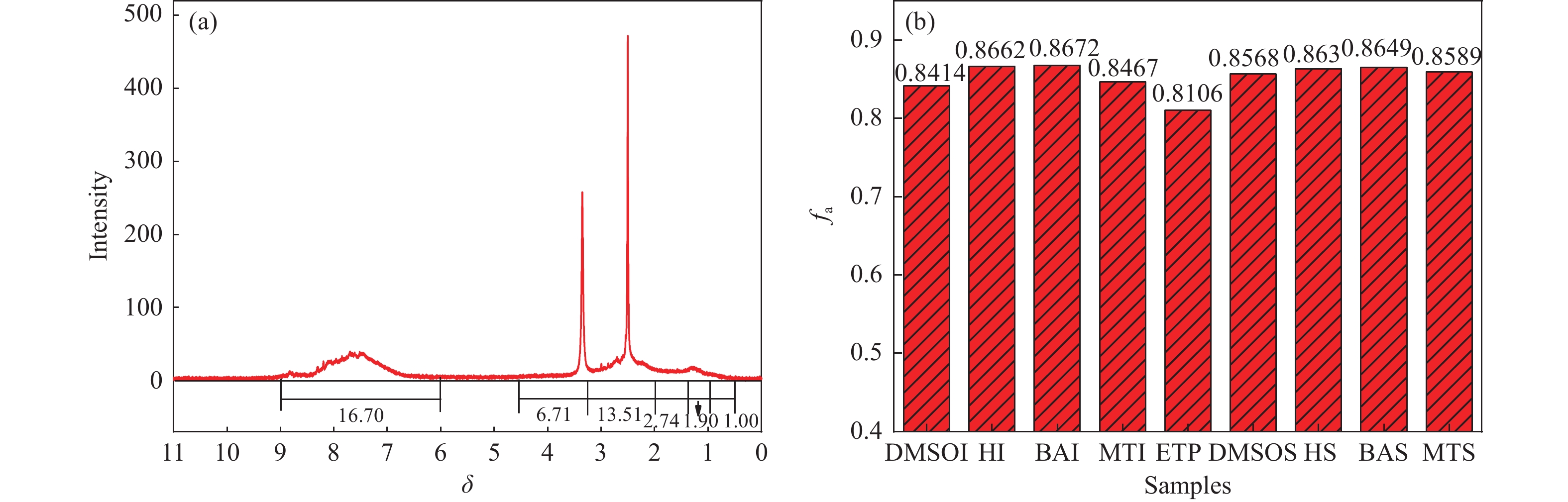
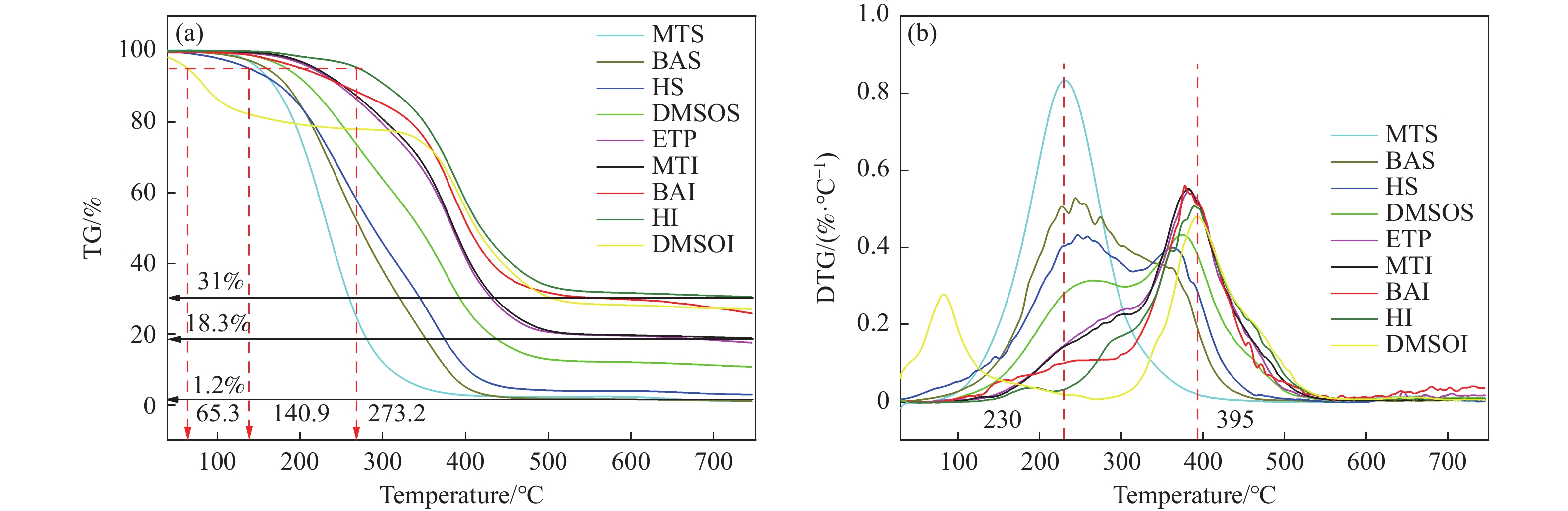
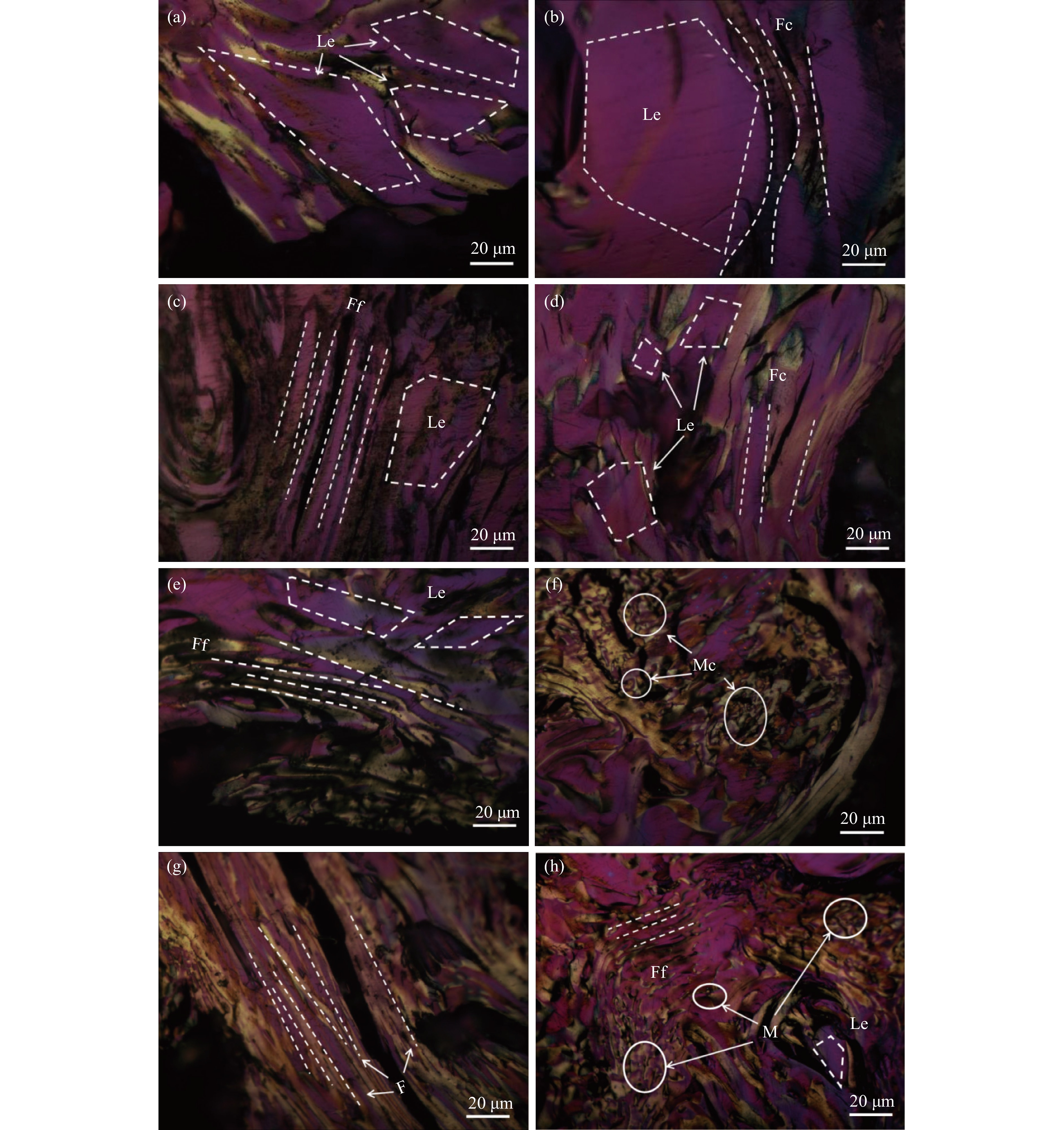
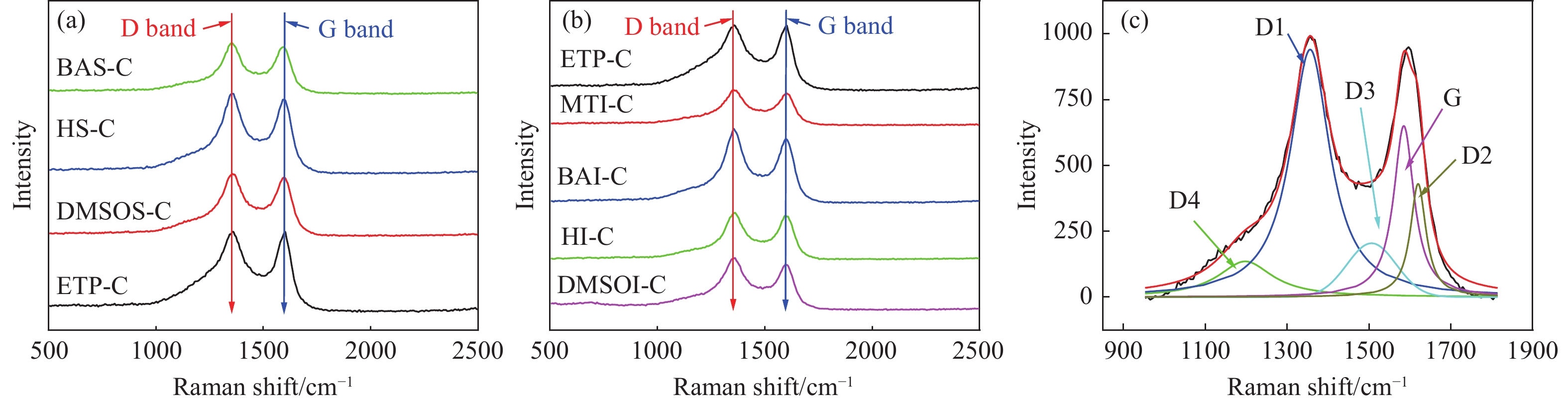
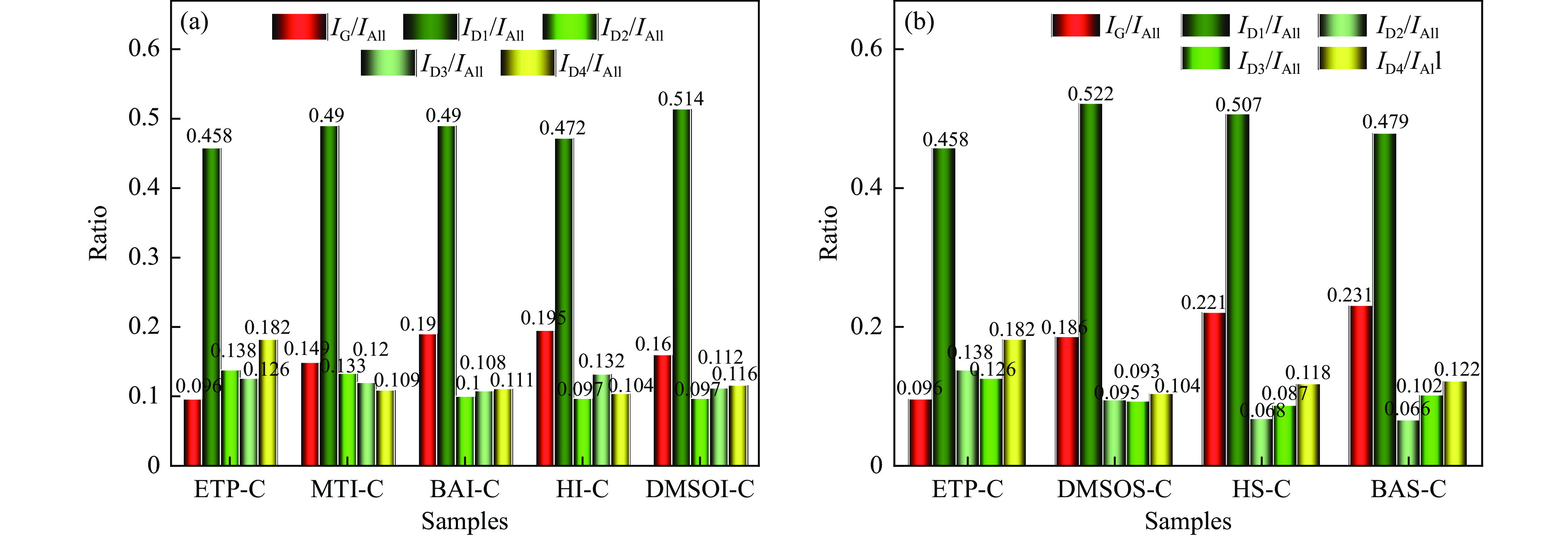
乙烯渣油沥青作为乙烯渣油中的重质组分具有含碳量高、芳香度较高及杂原子(S、N)含量低的特点,被广泛用作制备石油基人造炭材料的优选原料。为充分研究乙烯渣油沥青的成焦性,本研究选取四种溶剂(甲醇、正丁醇、正庚烷、二甲基亚砜)对乙烯渣油沥青进行萃取分离,获得八种乙烯渣油沥青组分(四种可溶组分和四种不溶组分),并对获得的乙烯渣油沥青组分进行热转化和炭化处理(热转化温度和炭化温度分别为500和1400 ℃)获得系列乙烯渣油沥青焦。采用红外光谱、热重分析仪、1H-NMR对乙烯渣油沥青组分的基础物性进行研究;利用偏光显微镜、X射线单晶衍射仪、拉曼光谱仪、扫描电子显微镜等对系列石油系沥青焦结构进行研究。结果表明,乙烯渣油沥青不溶组分芳香性略高于可溶组分,且不溶组分支链略少于可溶组分;不溶组分热转化和炭化后得到的乙烯渣油沥青焦显微强度高于可溶组分获得的乙烯渣油沥青焦,且乙烯渣油沥青沥青焦HS-C的真密度高达2.0554 g/cm3。
乙烯渣油沥青作为乙烯渣油中的重质组分具有含碳量高、芳香度较高及杂原子(S、N)含量低的特点,被广泛用作制备石油基人造炭材料的优选原料。为充分研究乙烯渣油沥青的成焦性,本研究选取四种溶剂(甲醇、正丁醇、正庚烷、二甲基亚砜)对乙烯渣油沥青进行萃取分离,获得八种乙烯渣油沥青组分(四种可溶组分和四种不溶组分),并对获得的乙烯渣油沥青组分进行热转化和炭化处理(热转化温度和炭化温度分别为500和1400 ℃)获得系列乙烯渣油沥青焦。采用红外光谱、热重分析仪、1H-NMR对乙烯渣油沥青组分的基础物性进行研究;利用偏光显微镜、X射线单晶衍射仪、拉曼光谱仪、扫描电子显微镜等对系列石油系沥青焦结构进行研究。结果表明,乙烯渣油沥青不溶组分芳香性略高于可溶组分,且不溶组分支链略少于可溶组分;不溶组分热转化和炭化后得到的乙烯渣油沥青焦显微强度高于可溶组分获得的乙烯渣油沥青焦,且乙烯渣油沥青沥青焦HS-C的真密度高达2.0554 g/cm3。





当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60445-7
摘要:
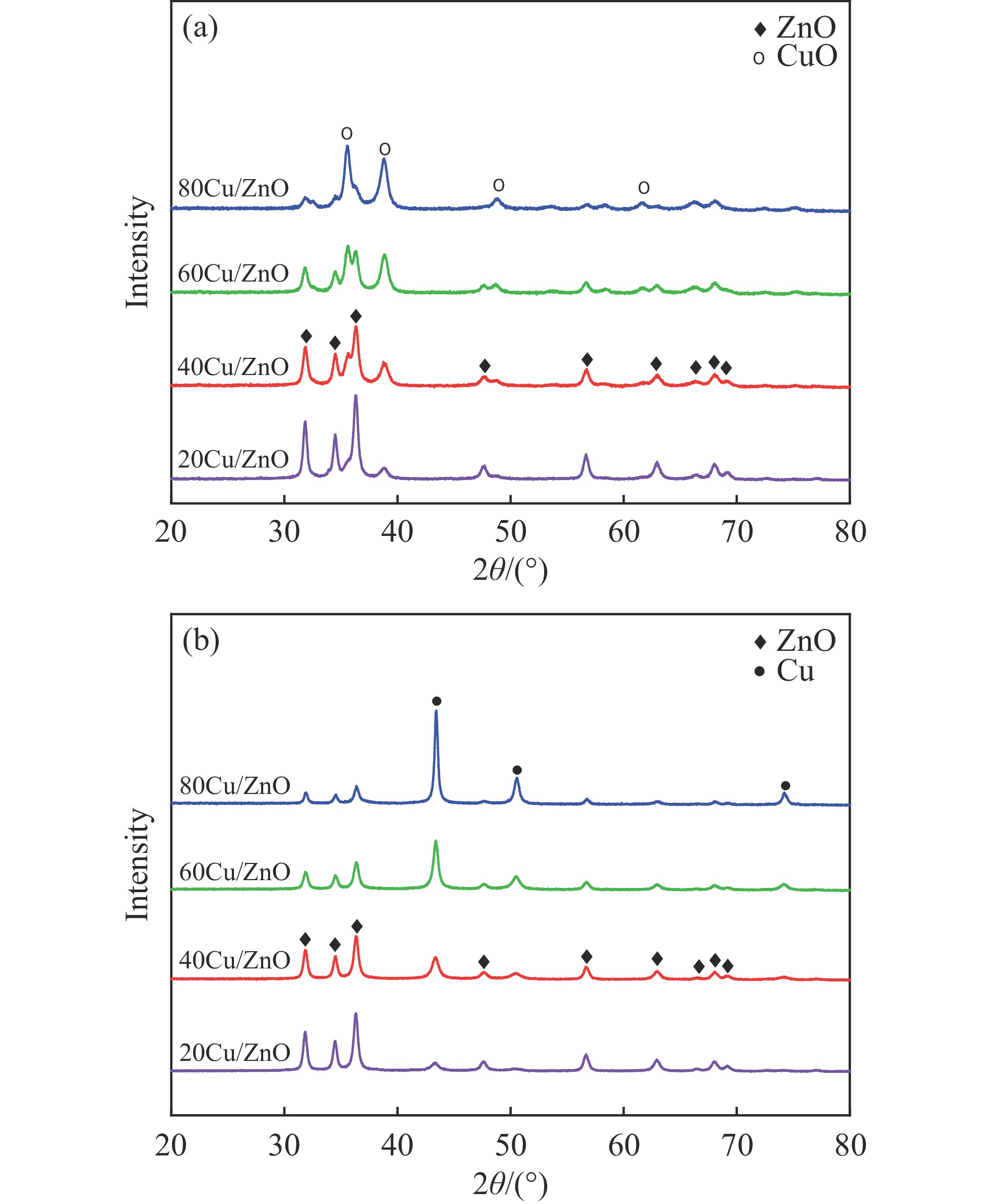
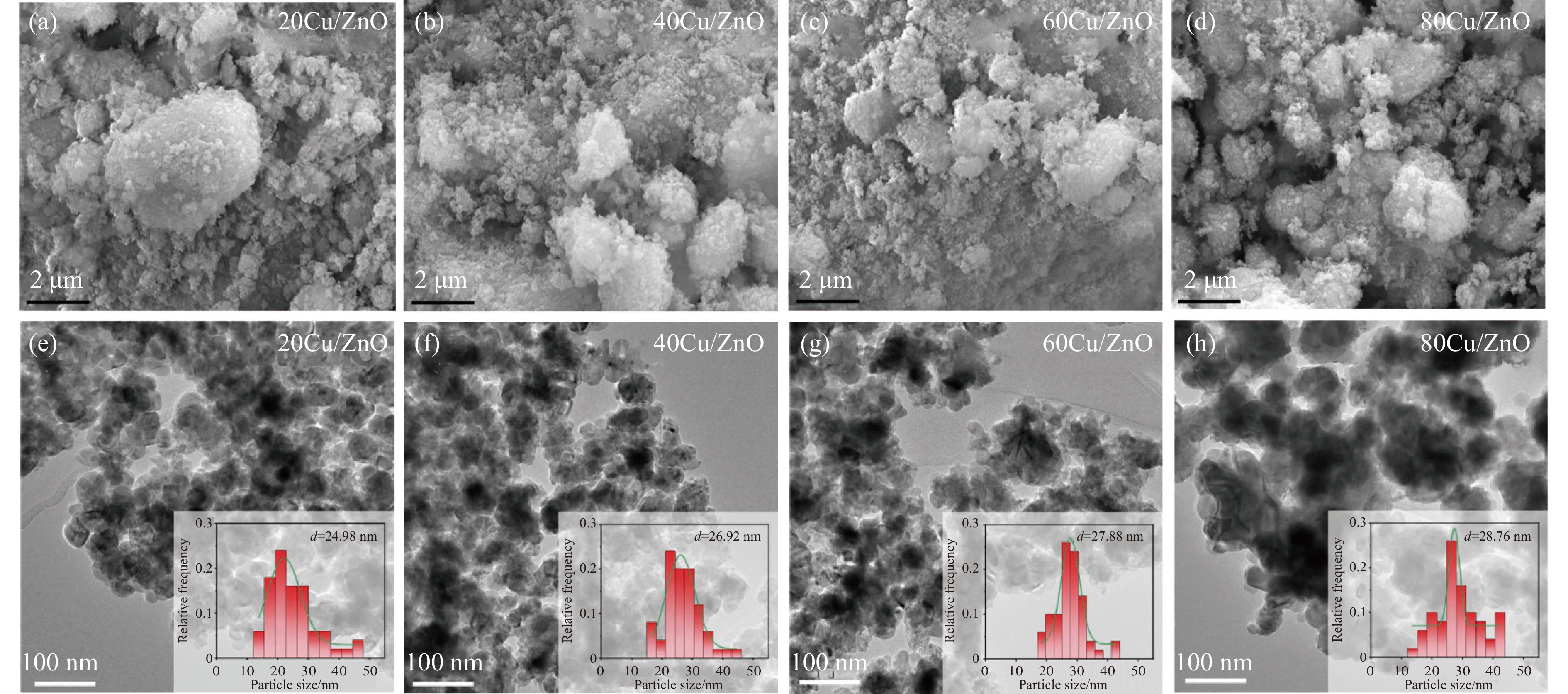
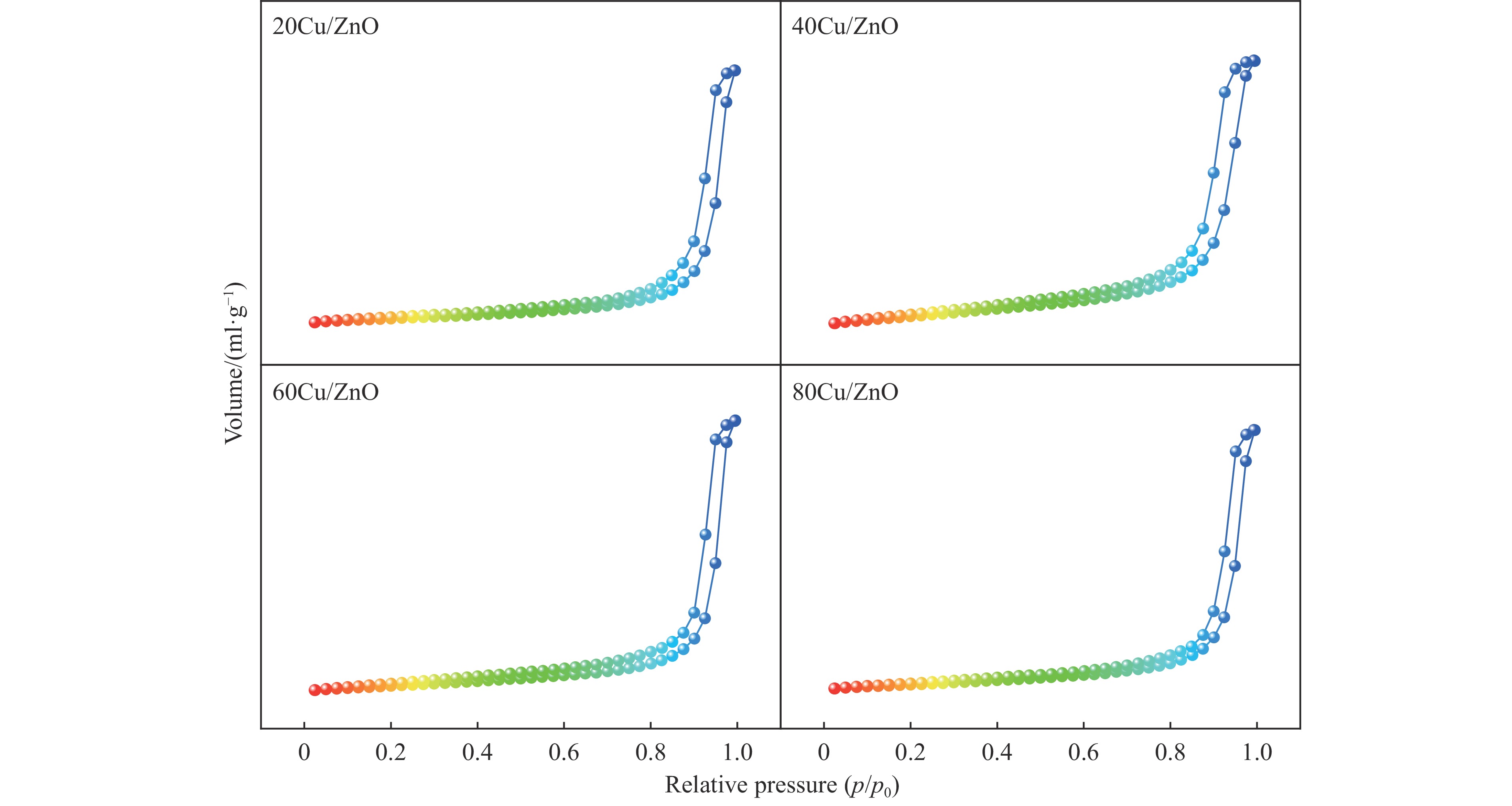
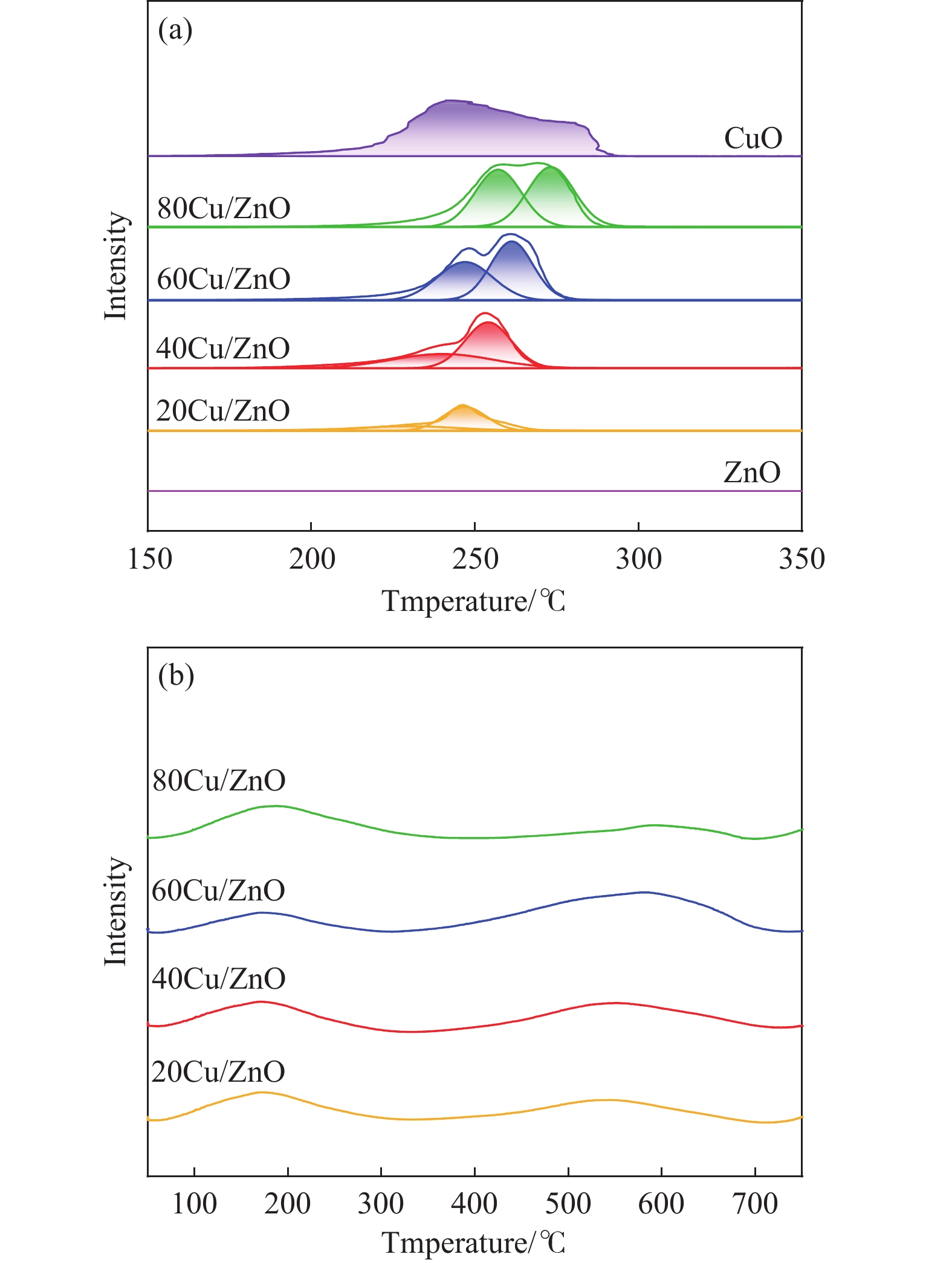
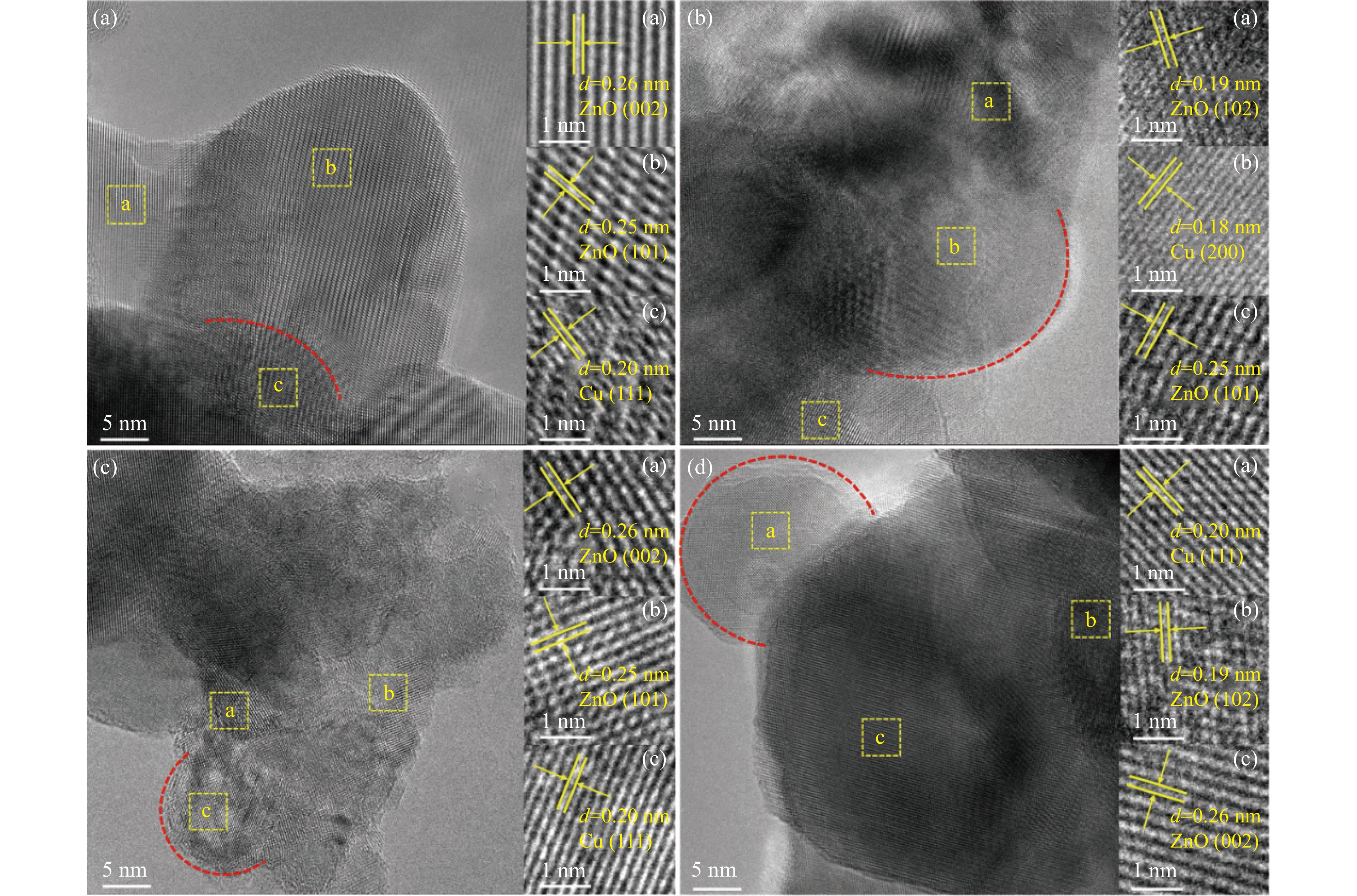
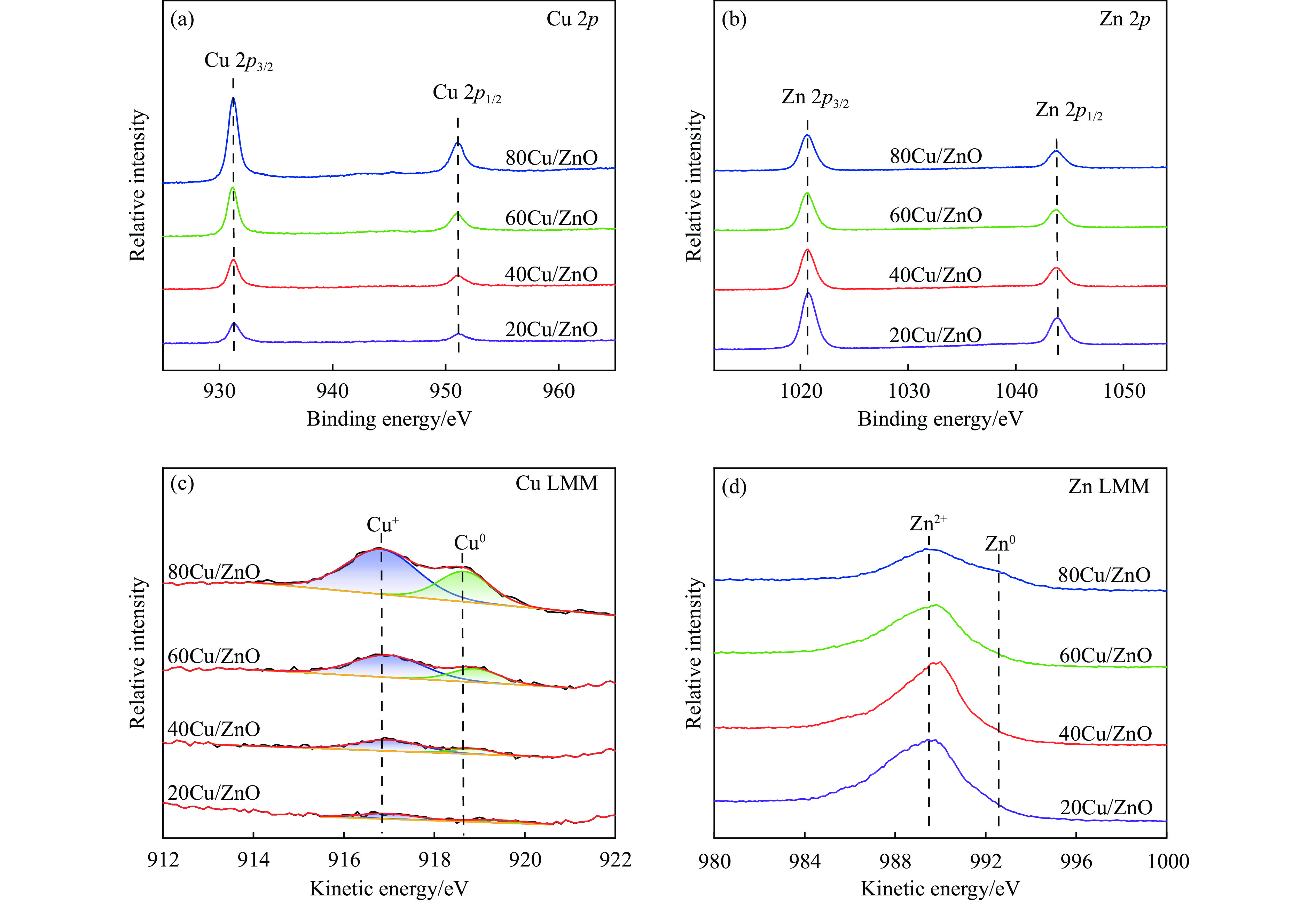
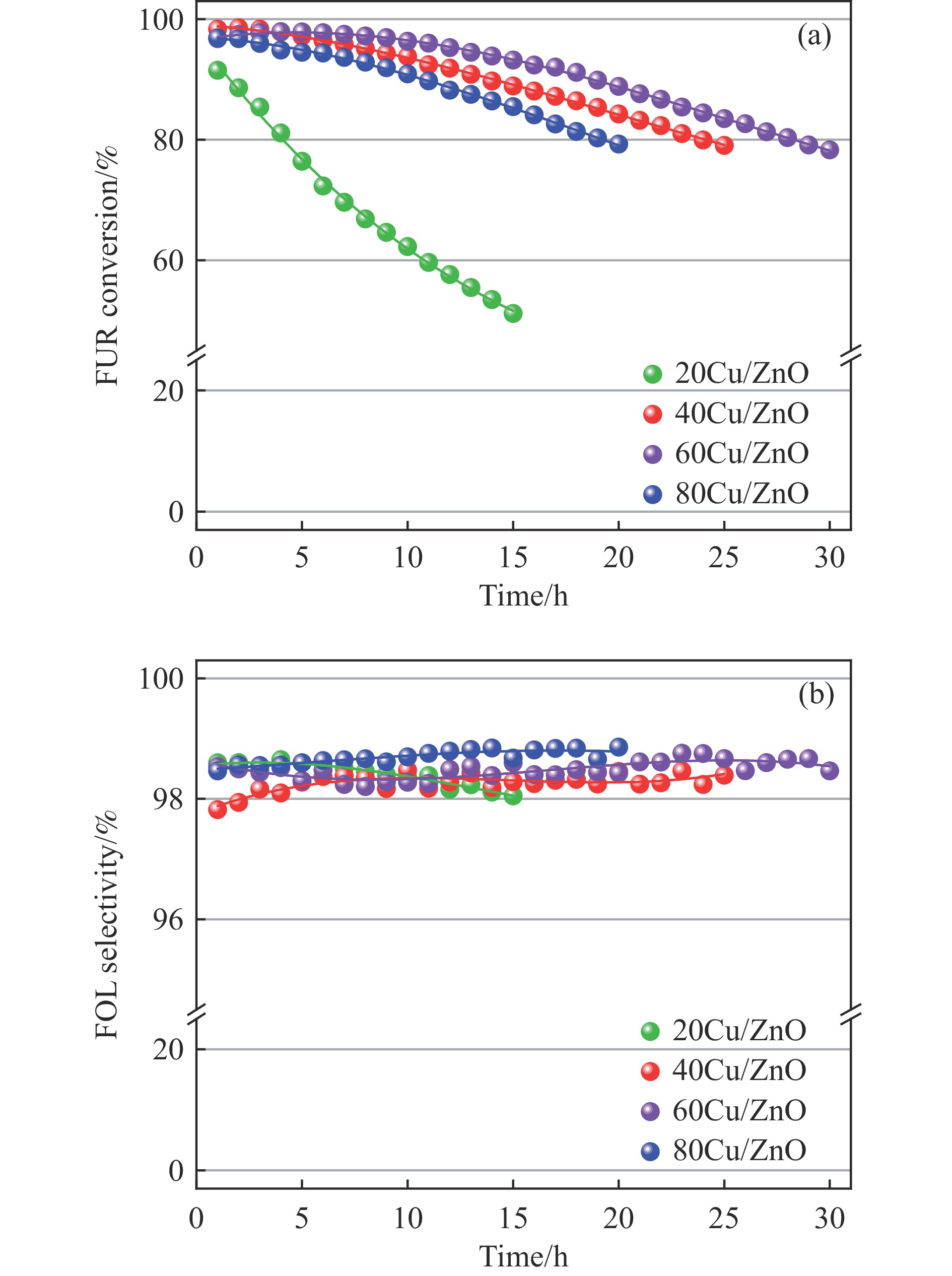
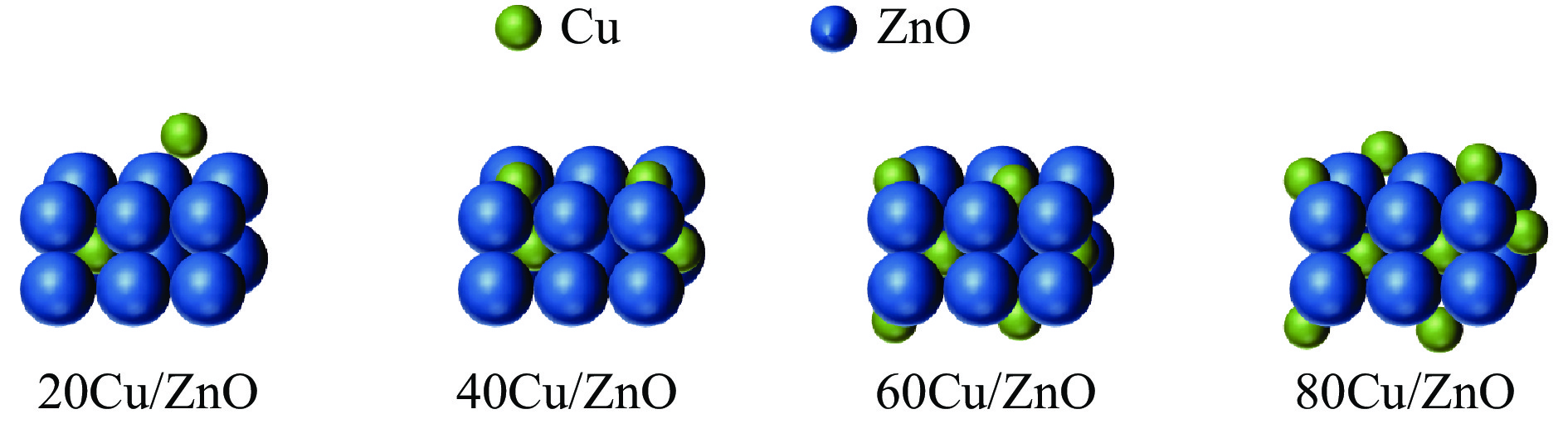
本研究考察了共沉淀法制备的Cu/ZnO催化剂中Cu/Zn比例对金属载体强相互作用以及其催化糠醛气相加氢制糠醇反应性能的影响。XRD、H2-TPR、SEM、HRTEM、XPS表征结果表明,Cu/ZnO催化剂体系中存在金属载体强相互作用(SMSI),改变了催化剂的微观结构。ZnO载体对活性金属Cu颗粒形成了不同程度的几何修饰,影响了Cu表面电子状态。Cu/Zn比例影响催化剂SMSI作用,SMSI作用顺序是20Cu/ZnO> 40Cu/ZnO> 60Cu/ZnO> 80Cu/ZnO。在同一反应条件下,20Cu/ZnO催化剂的糠醛转化率高于80%的时间仅为5 h,而60Cu/ZnO催化剂的糠醛转化率高于80%的时间可以达到28 h,表明过强的SMSI作用会抑制催化剂的活性,适当的SMSI作用使Cu/ZnO催化剂在糠醛加氢反应中的稳定性得到提升。
本研究考察了共沉淀法制备的Cu/ZnO催化剂中Cu/Zn比例对金属载体强相互作用以及其催化糠醛气相加氢制糠醇反应性能的影响。XRD、H2-TPR、SEM、HRTEM、XPS表征结果表明,Cu/ZnO催化剂体系中存在金属载体强相互作用(SMSI),改变了催化剂的微观结构。ZnO载体对活性金属Cu颗粒形成了不同程度的几何修饰,影响了Cu表面电子状态。Cu/Zn比例影响催化剂SMSI作用,SMSI作用顺序是20Cu/ZnO> 40Cu/ZnO> 60Cu/ZnO> 80Cu/ZnO。在同一反应条件下,20Cu/ZnO催化剂的糠醛转化率高于80%的时间仅为5 h,而60Cu/ZnO催化剂的糠醛转化率高于80%的时间可以达到28 h,表明过强的SMSI作用会抑制催化剂的活性,适当的SMSI作用使Cu/ZnO催化剂在糠醛加氢反应中的稳定性得到提升。

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024014
摘要:
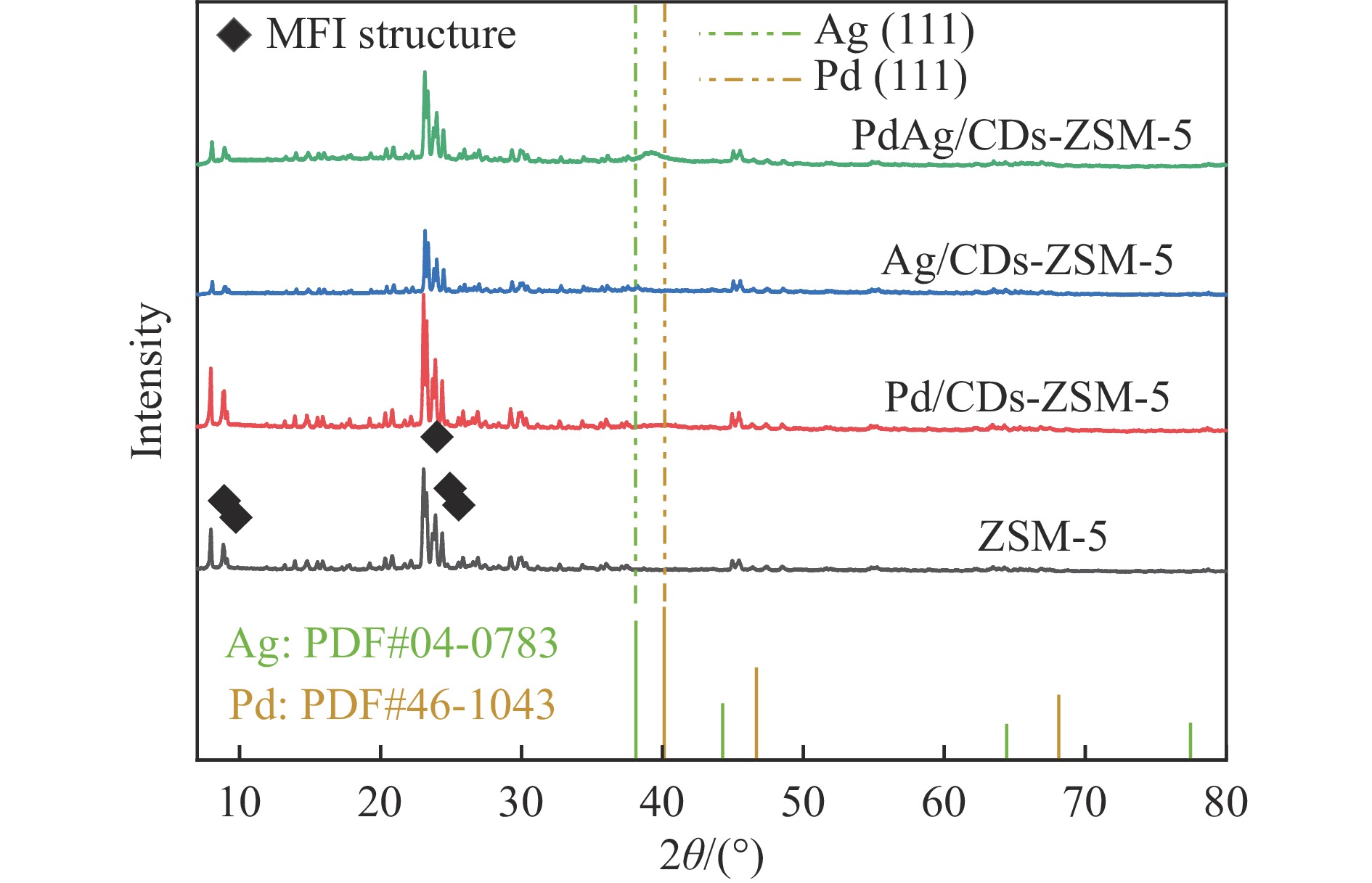
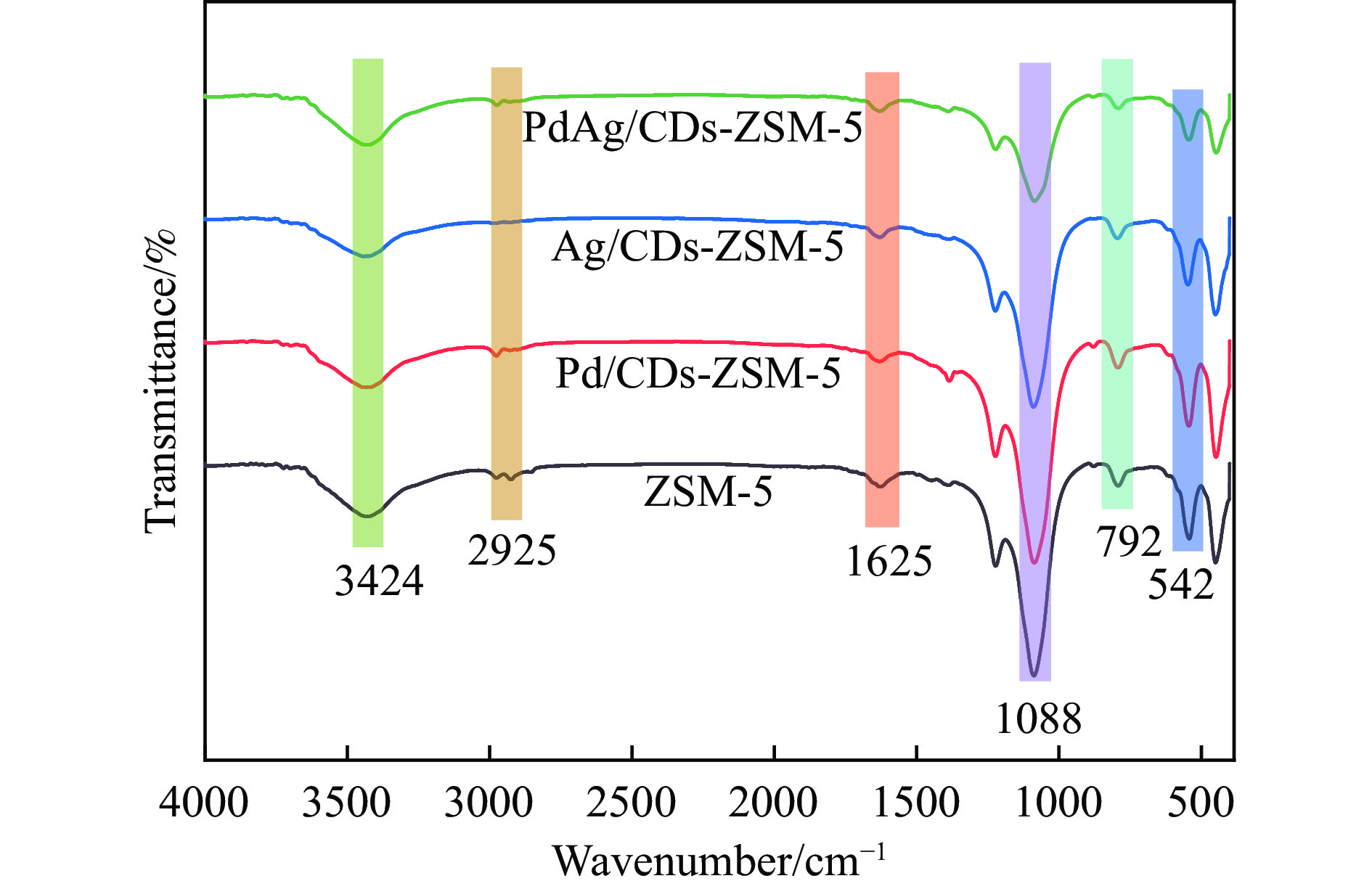
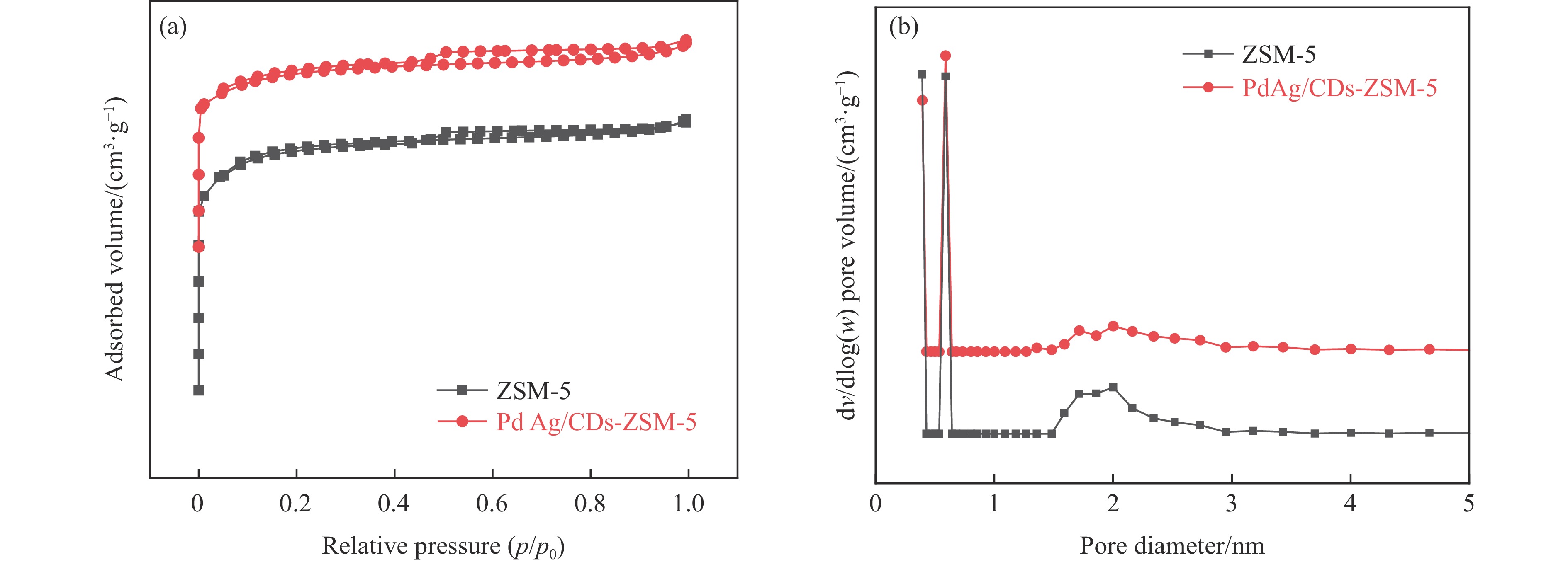
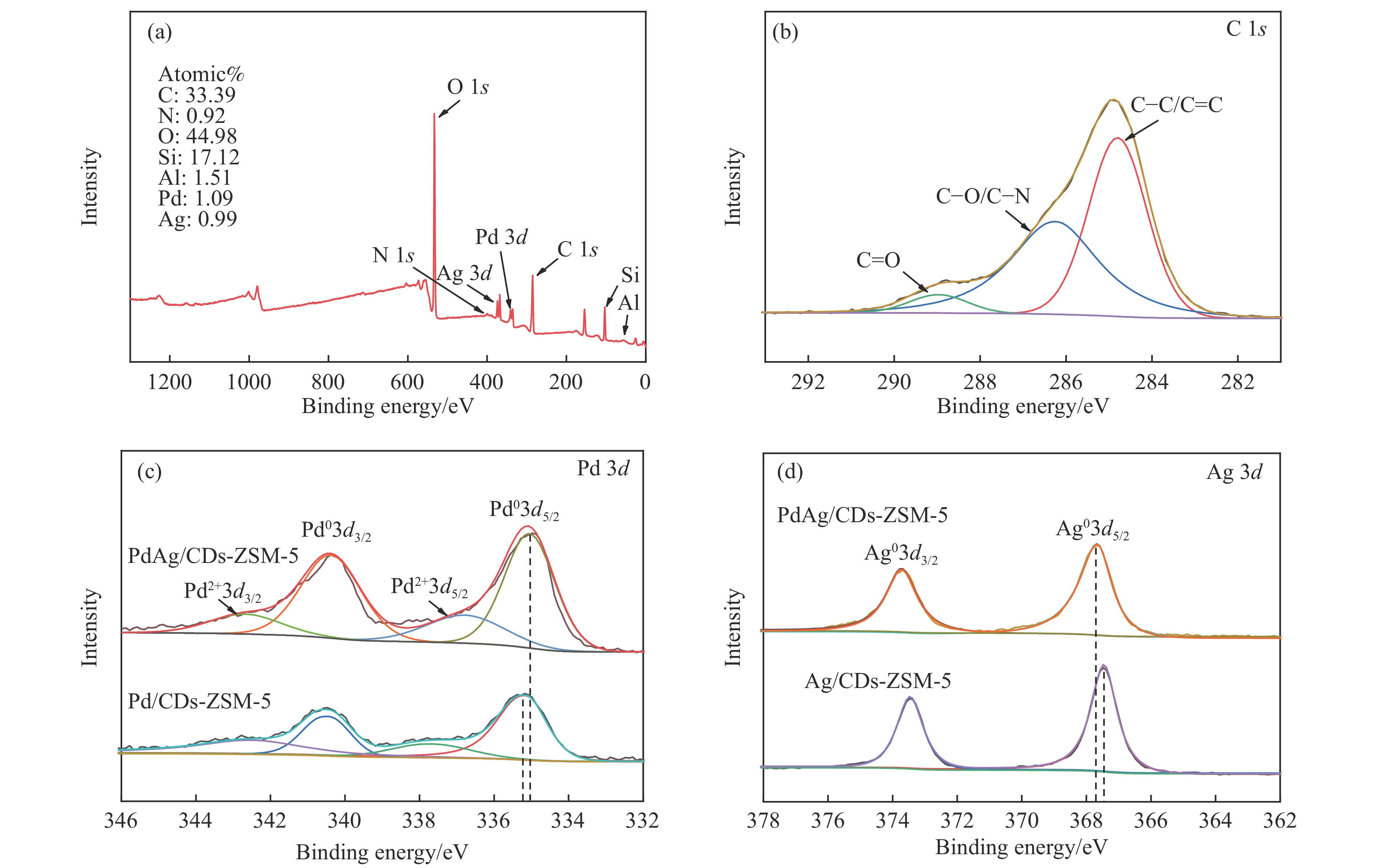
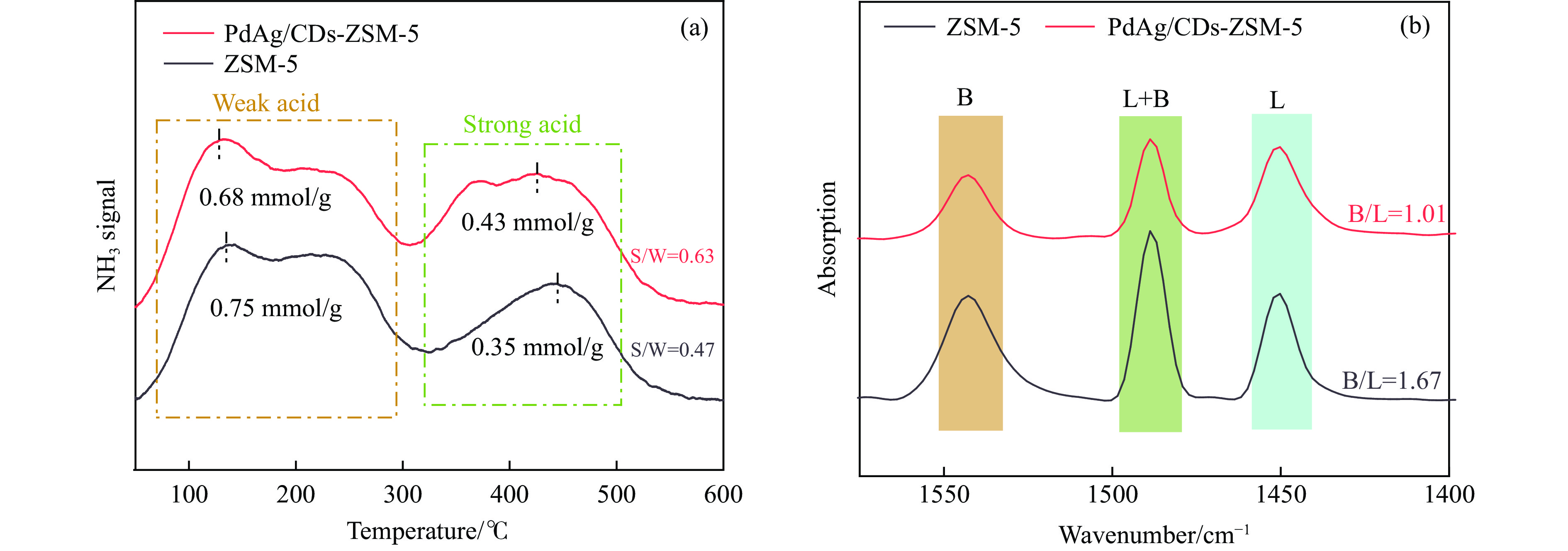
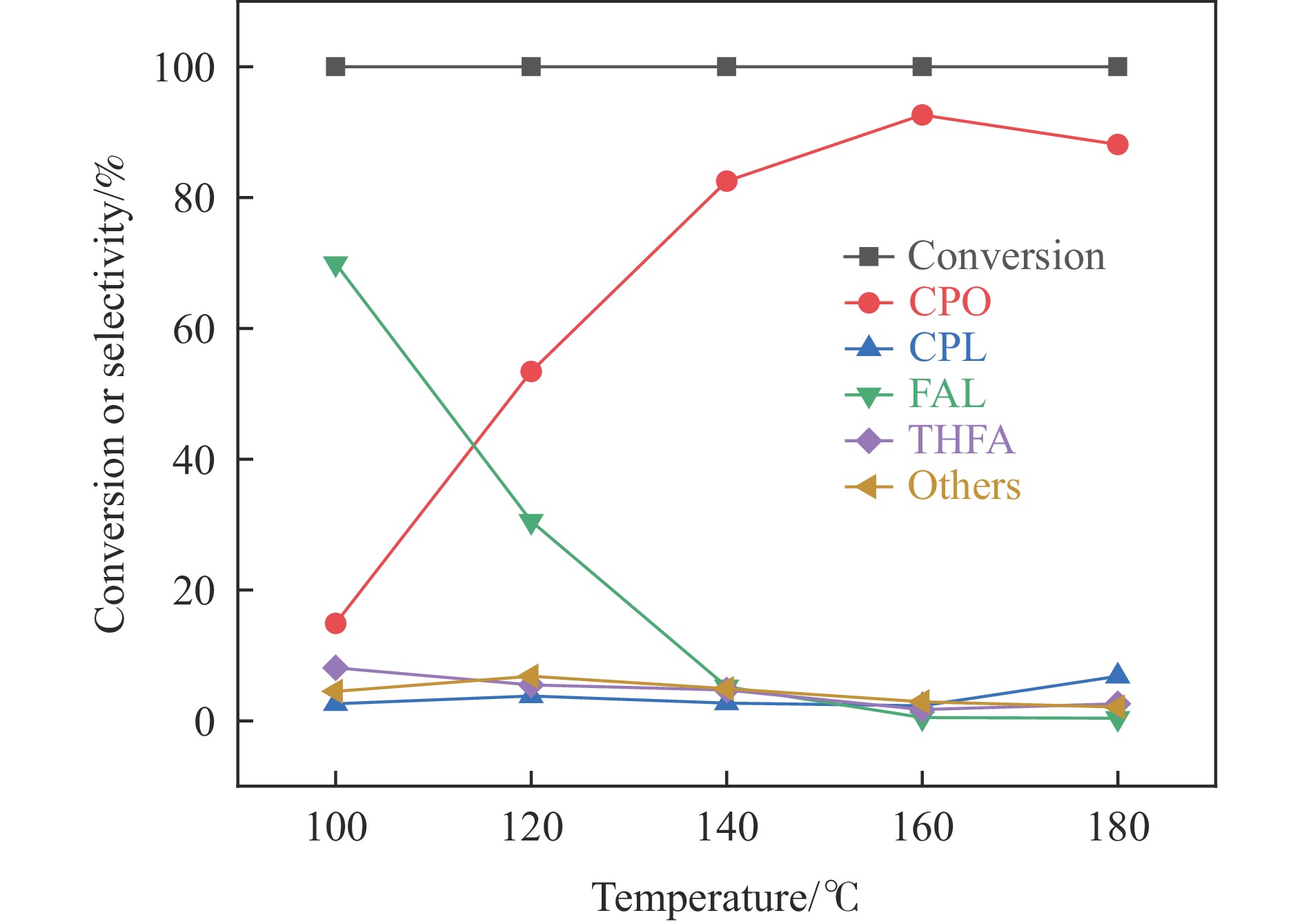
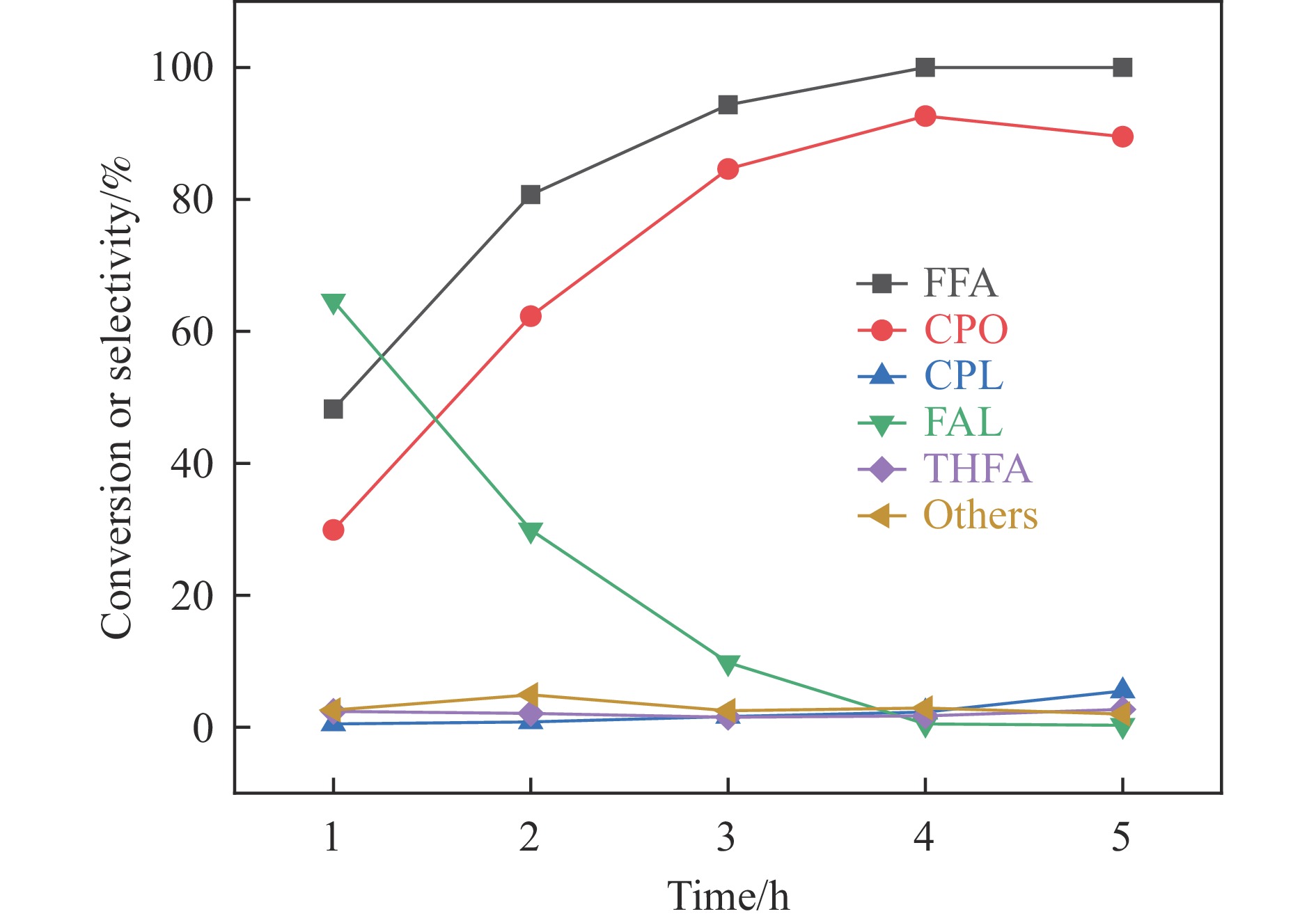
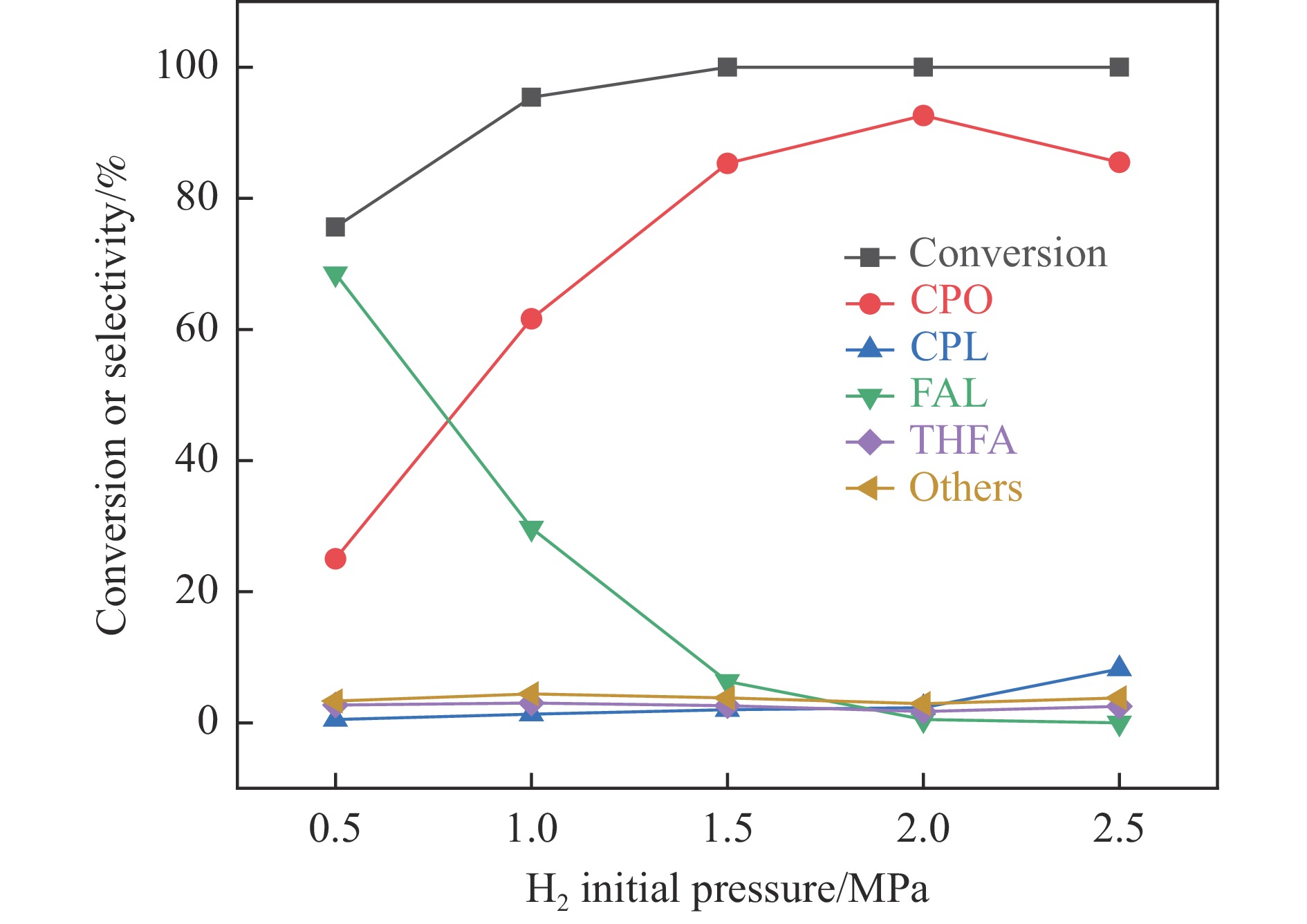
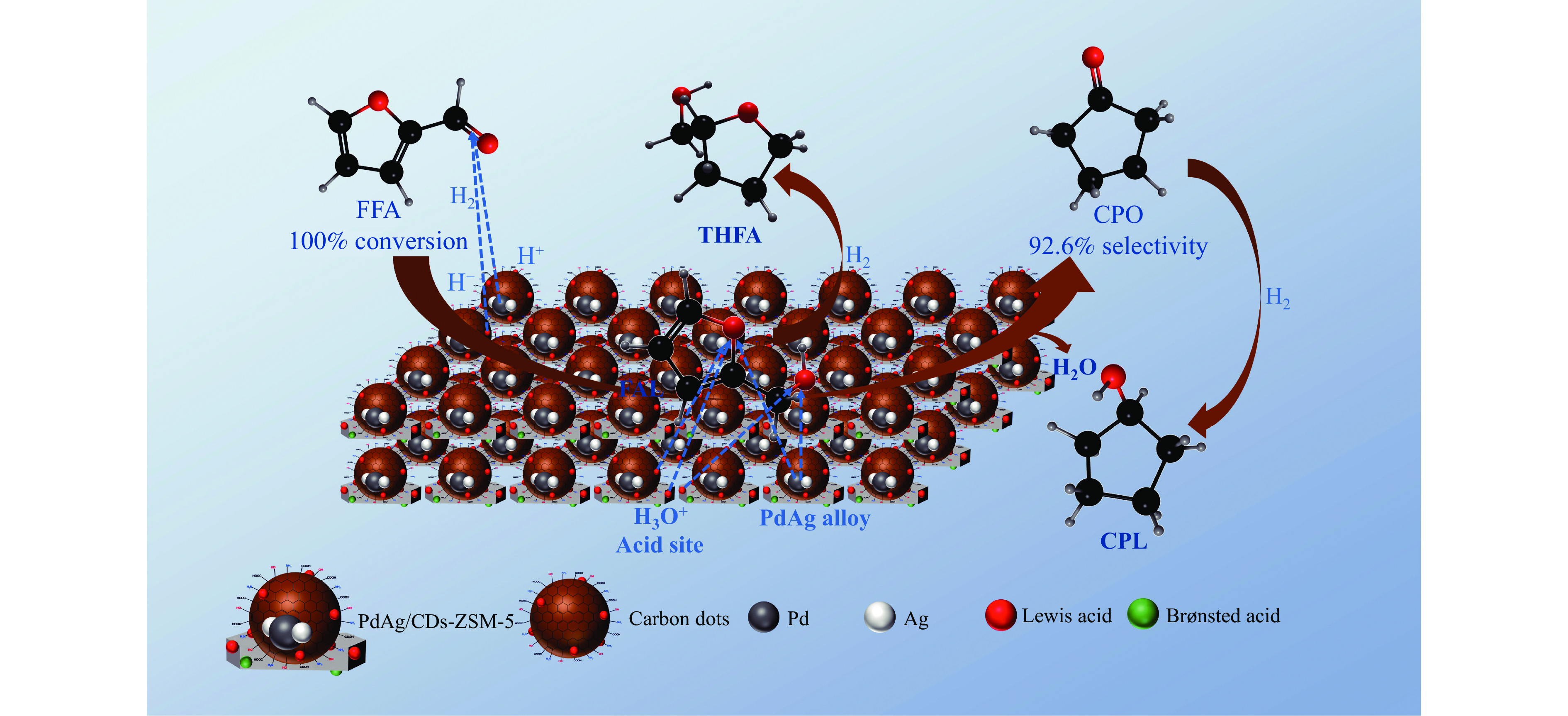
以沸石分子筛(ZSM-5)为载体,碳点(carbon dots, CDs)为还原剂和稳定剂,通过光照还原法制备了双金属PdAg/CDs-ZSM-5催化剂,用于糠醛(furfural, FFA)水相加氢-重排制备环戊酮(cyclopentanone, CPO)反应。采用X射线衍射(XRD)、X射线光电子能谱(XPS)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、氨气程序化学吸附(NH3-TPD)和吡啶红外(Py-FTIR)等手段对催化剂进行了表征。结果表明,CDs具有良好的还原性和丰富的Lewis酸性位点,能够将Pd2+、Ag+还原为金属单质并形成纳米合金结构,复合催化剂中适宜的酸性位点与PdAg合金之间的协同作用使得PdAg/CDs-ZSM-5催化剂在最优反应条件下,对FFA转化率达到100%,目标产物CPO选择性为92.6%。催化剂重复使用五次后仍能保持较高的活性与稳定性。
以沸石分子筛(ZSM-5)为载体,碳点(carbon dots, CDs)为还原剂和稳定剂,通过光照还原法制备了双金属PdAg/CDs-ZSM-5催化剂,用于糠醛(furfural, FFA)水相加氢-重排制备环戊酮(cyclopentanone, CPO)反应。采用X射线衍射(XRD)、X射线光电子能谱(XPS)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、氨气程序化学吸附(NH3-TPD)和吡啶红外(Py-FTIR)等手段对催化剂进行了表征。结果表明,CDs具有良好的还原性和丰富的Lewis酸性位点,能够将Pd2+、Ag+还原为金属单质并形成纳米合金结构,复合催化剂中适宜的酸性位点与PdAg合金之间的协同作用使得PdAg/CDs-ZSM-5催化剂在最优反应条件下,对FFA转化率达到100%,目标产物CPO选择性为92.6%。催化剂重复使用五次后仍能保持较高的活性与稳定性。




当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60442-1
摘要:
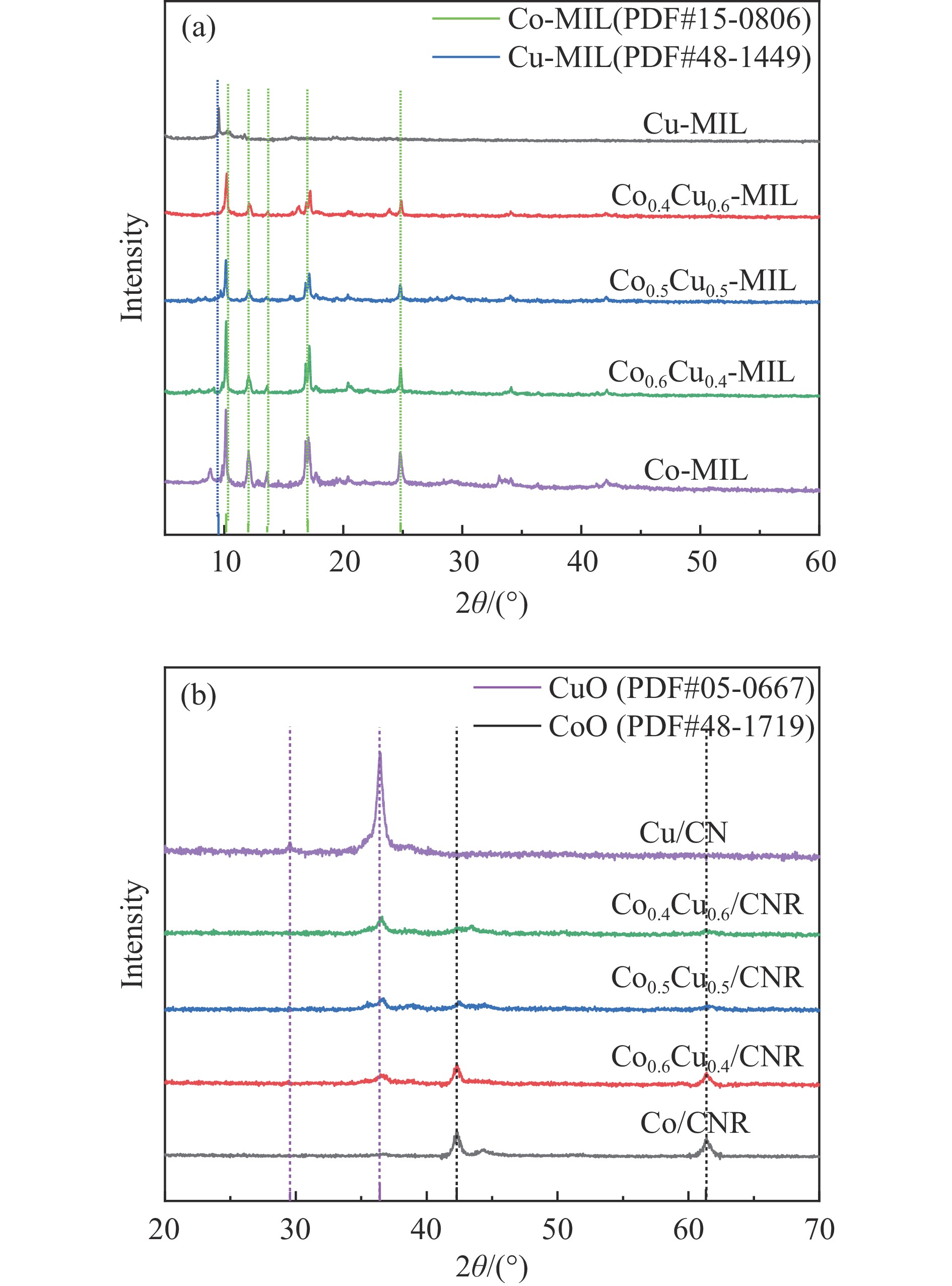
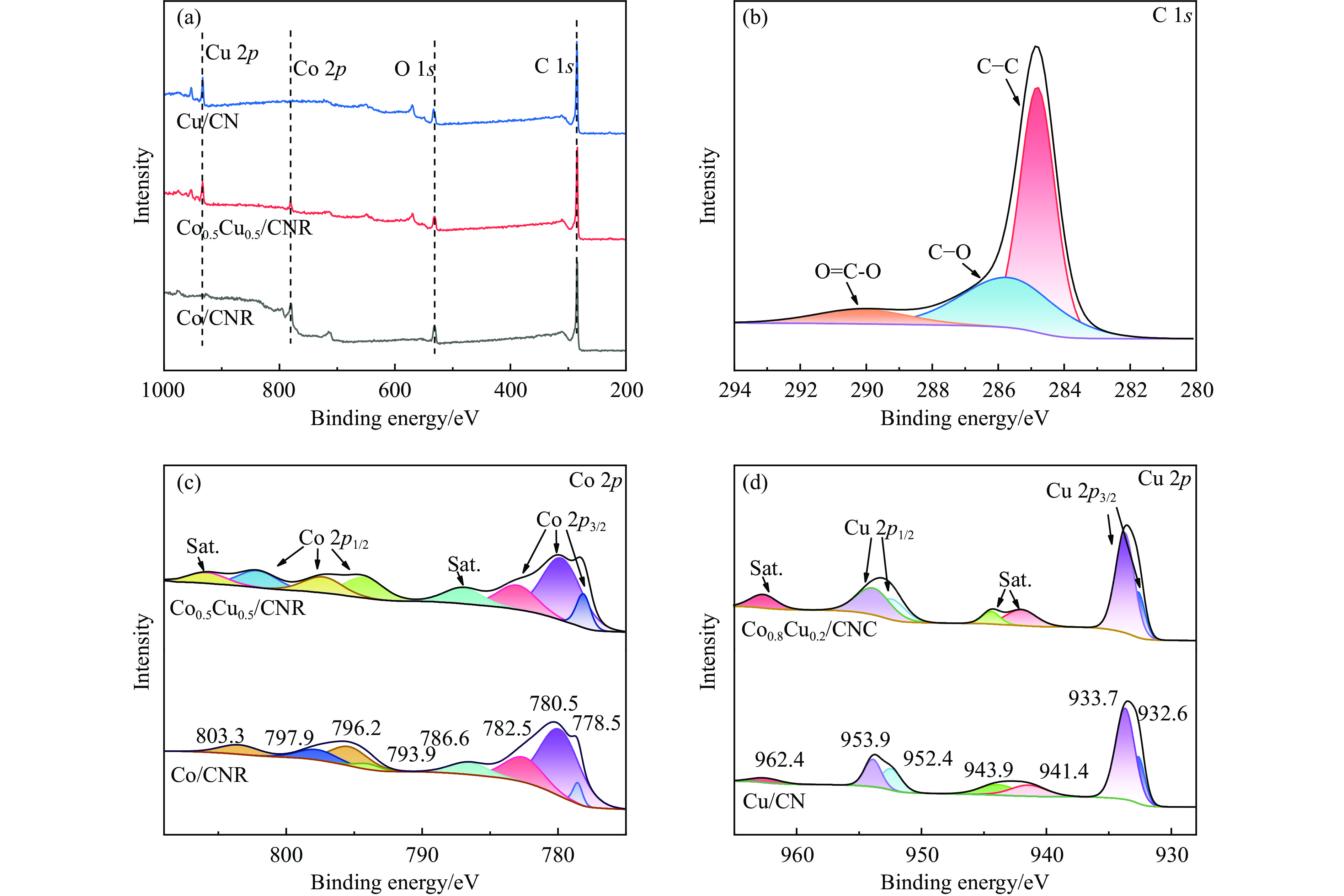
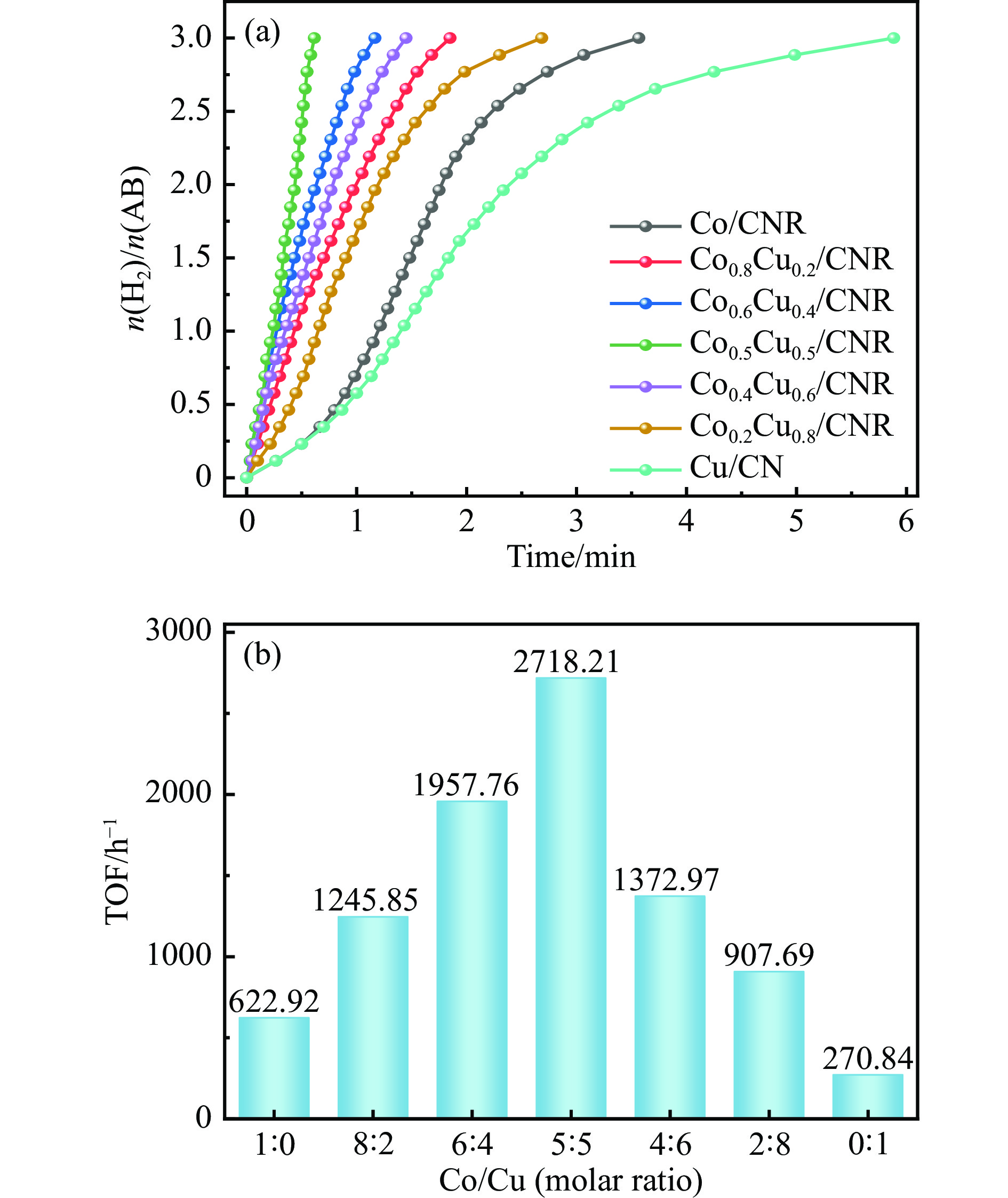
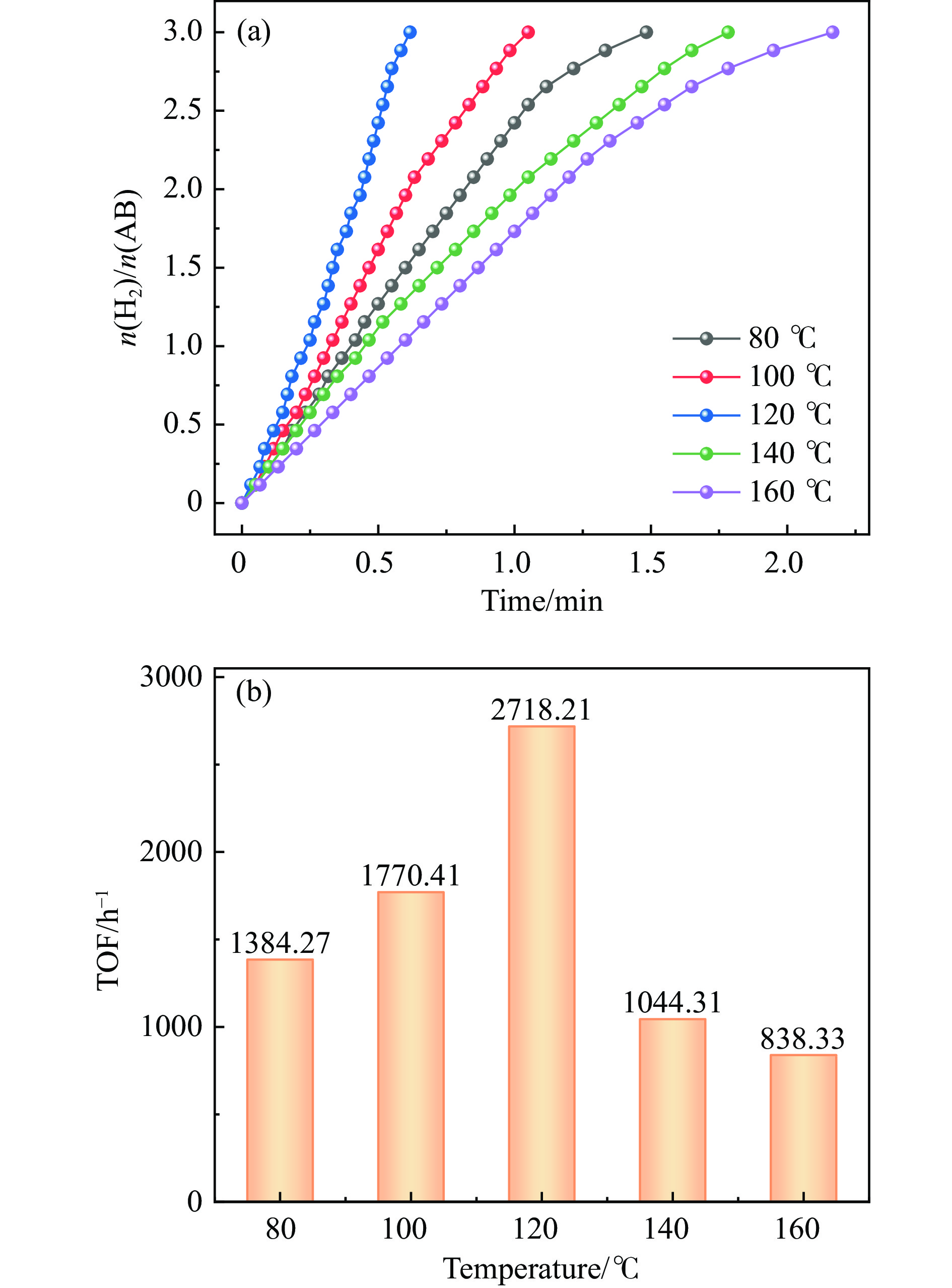
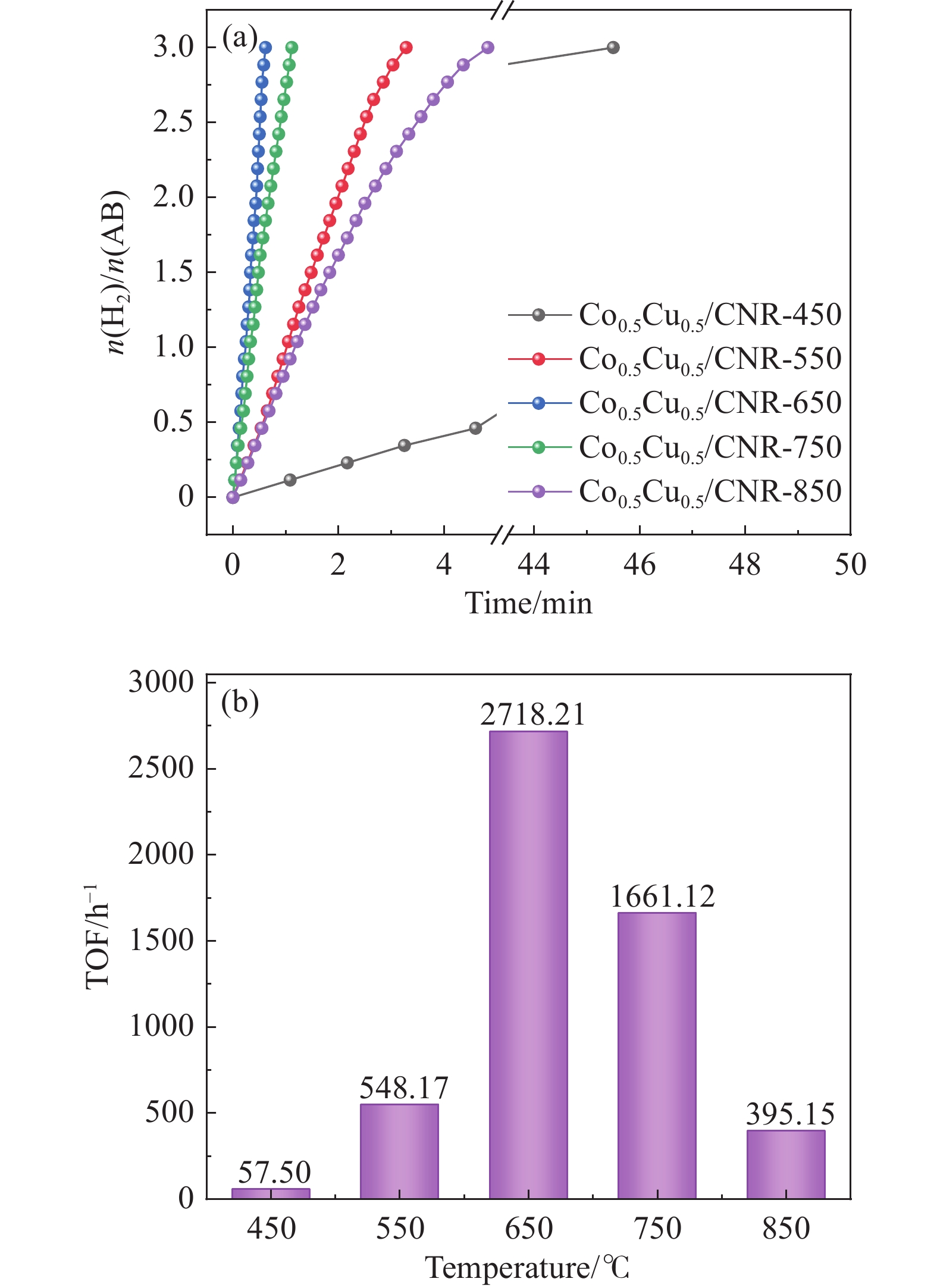
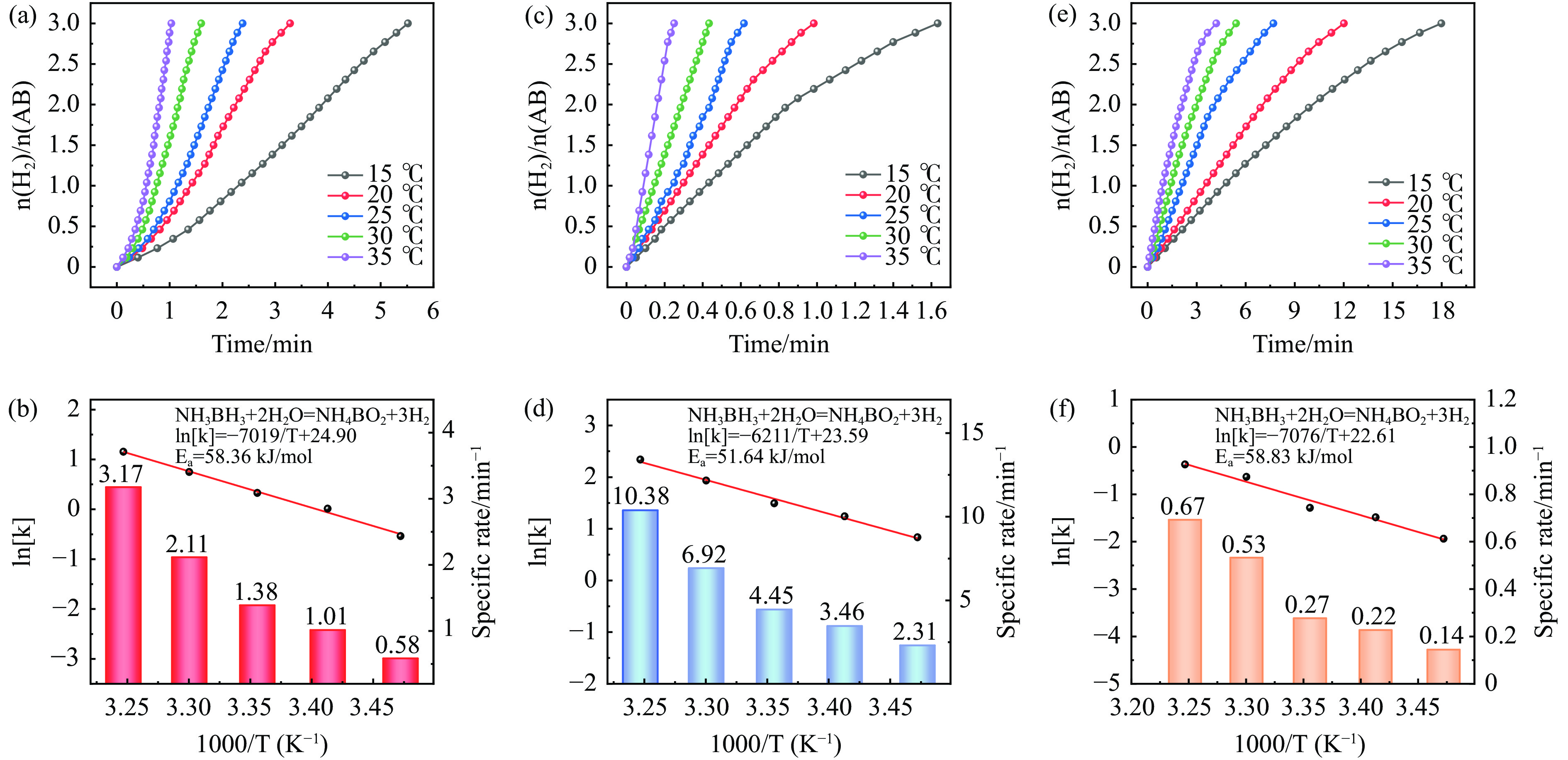
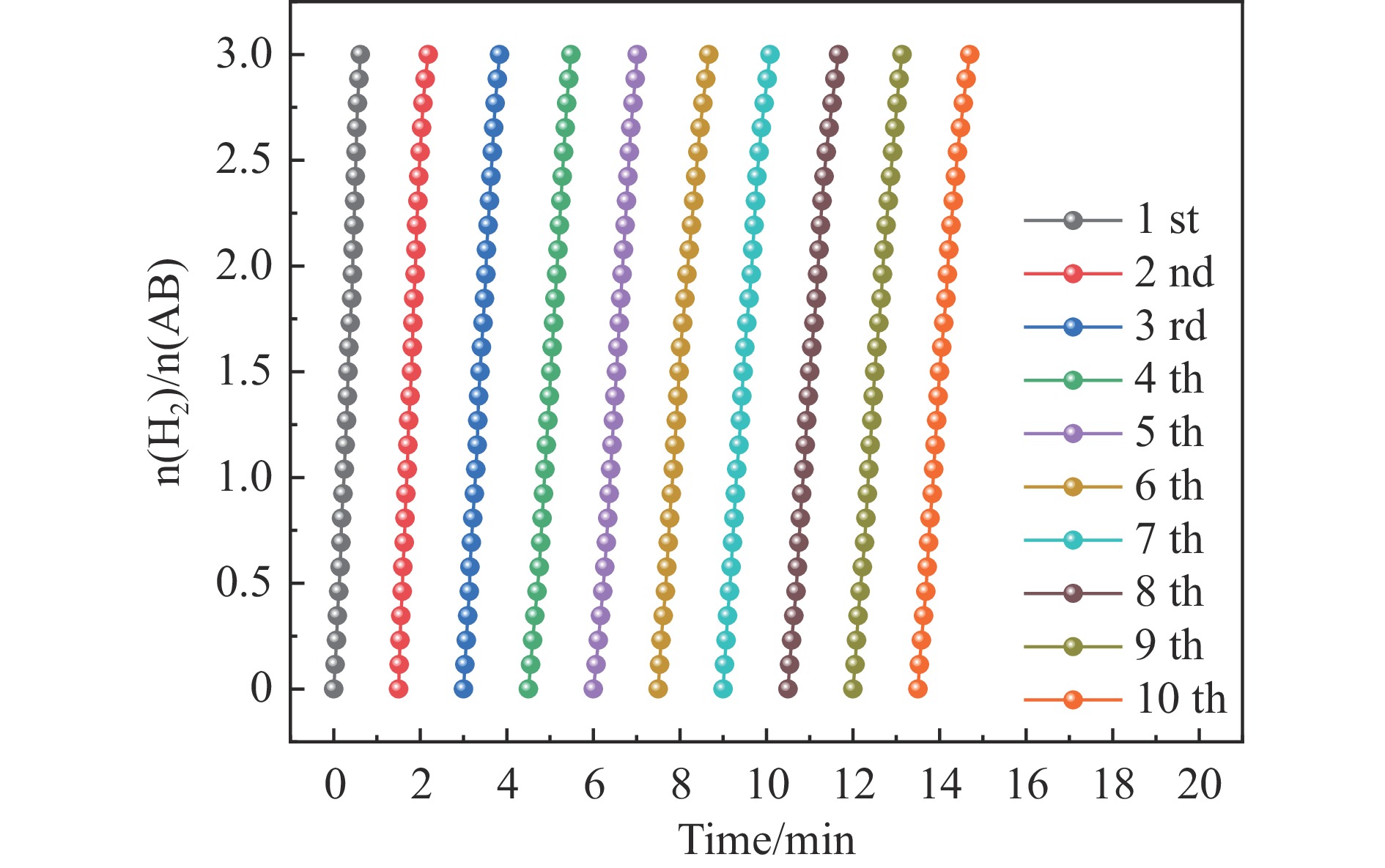
以硝酸钴和硝酸铜制备溶液A,苯二甲酸(PTA)和N,N-二甲基甲酰胺(DMF)制备溶液B,两种溶液通过溶剂热法制备Co/Cu拉瓦希尔骨架系列材料(Co/Cu-MIL前驱体),进一步直接碳化前驱体制备出MOFs衍生物,即双金属碳纳米棒(CoxCu1−x/CNR)催化剂。通过SEM、TEM、XRD、XPS等表征手段探究其形貌和组成。结果表明,Co/Cu-MIL经过高温焙烧后成功得到CoxCu1−x/CNR,当x=0.5、溶剂热温度为120 ℃、焙烧温度为650 ℃时得到的催化剂催化活性最优,Co0.5Cu0.5/CNR催化剂催化氨硼烷(AB)水解制氢的TOF值为2718.21 h−1,反应的活化能为51.64 kJ/mol,且催化剂的循环稳定性较好,在循环10次后催化活性虽然有所下降,但对AB仍然保持100%的转化率。
以硝酸钴和硝酸铜制备溶液A,苯二甲酸(PTA)和N,N-二甲基甲酰胺(DMF)制备溶液B,两种溶液通过溶剂热法制备Co/Cu拉瓦希尔骨架系列材料(Co/Cu-MIL前驱体),进一步直接碳化前驱体制备出MOFs衍生物,即双金属碳纳米棒(CoxCu1−x/CNR)催化剂。通过SEM、TEM、XRD、XPS等表征手段探究其形貌和组成。结果表明,Co/Cu-MIL经过高温焙烧后成功得到CoxCu1−x/CNR,当x=0.5、溶剂热温度为120 ℃、焙烧温度为650 ℃时得到的催化剂催化活性最优,Co0.5Cu0.5/CNR催化剂催化氨硼烷(AB)水解制氢的TOF值为2718.21 h−1,反应的活化能为51.64 kJ/mol,且催化剂的循环稳定性较好,在循环10次后催化活性虽然有所下降,但对AB仍然保持100%的转化率。



当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024010
摘要:
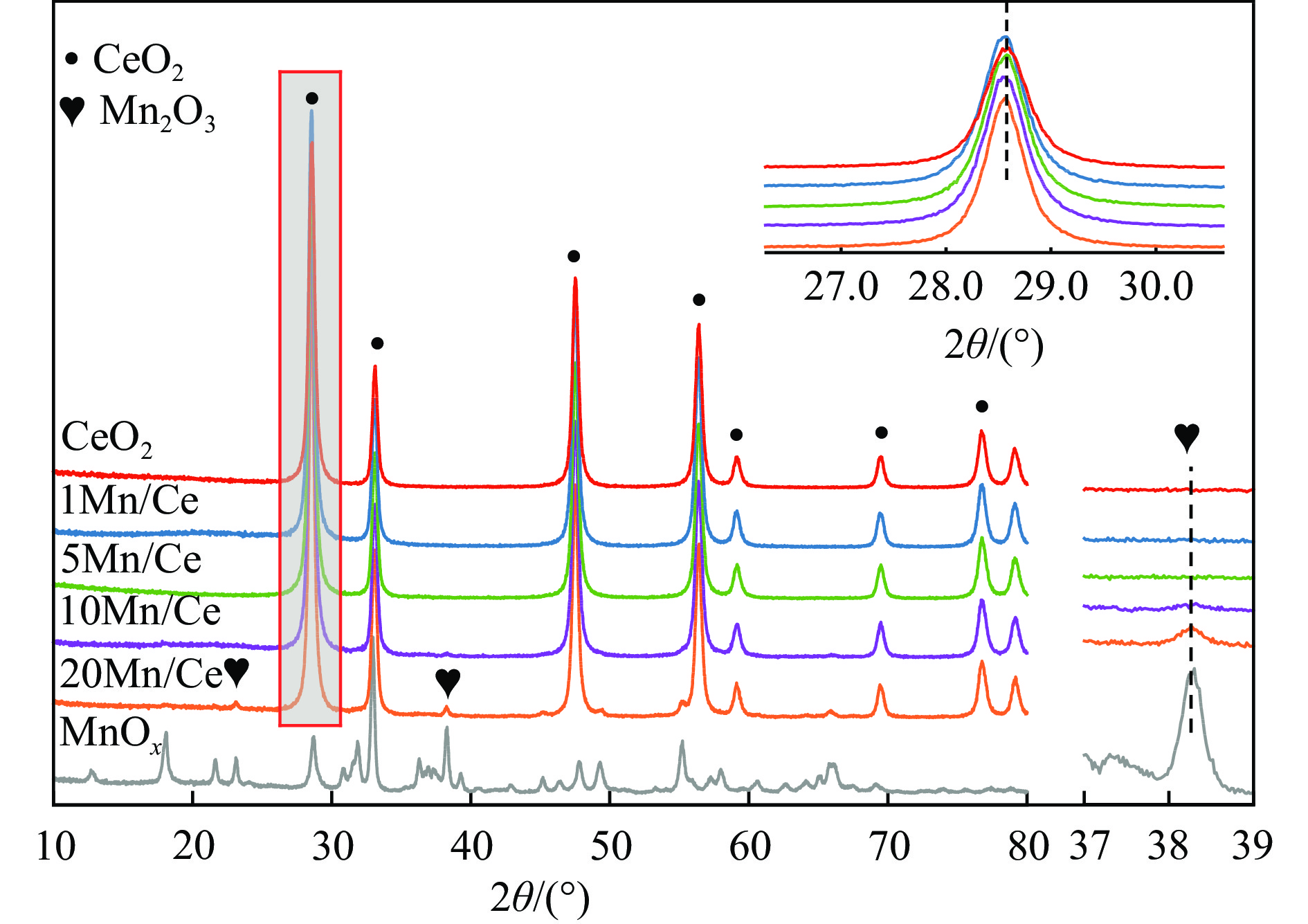
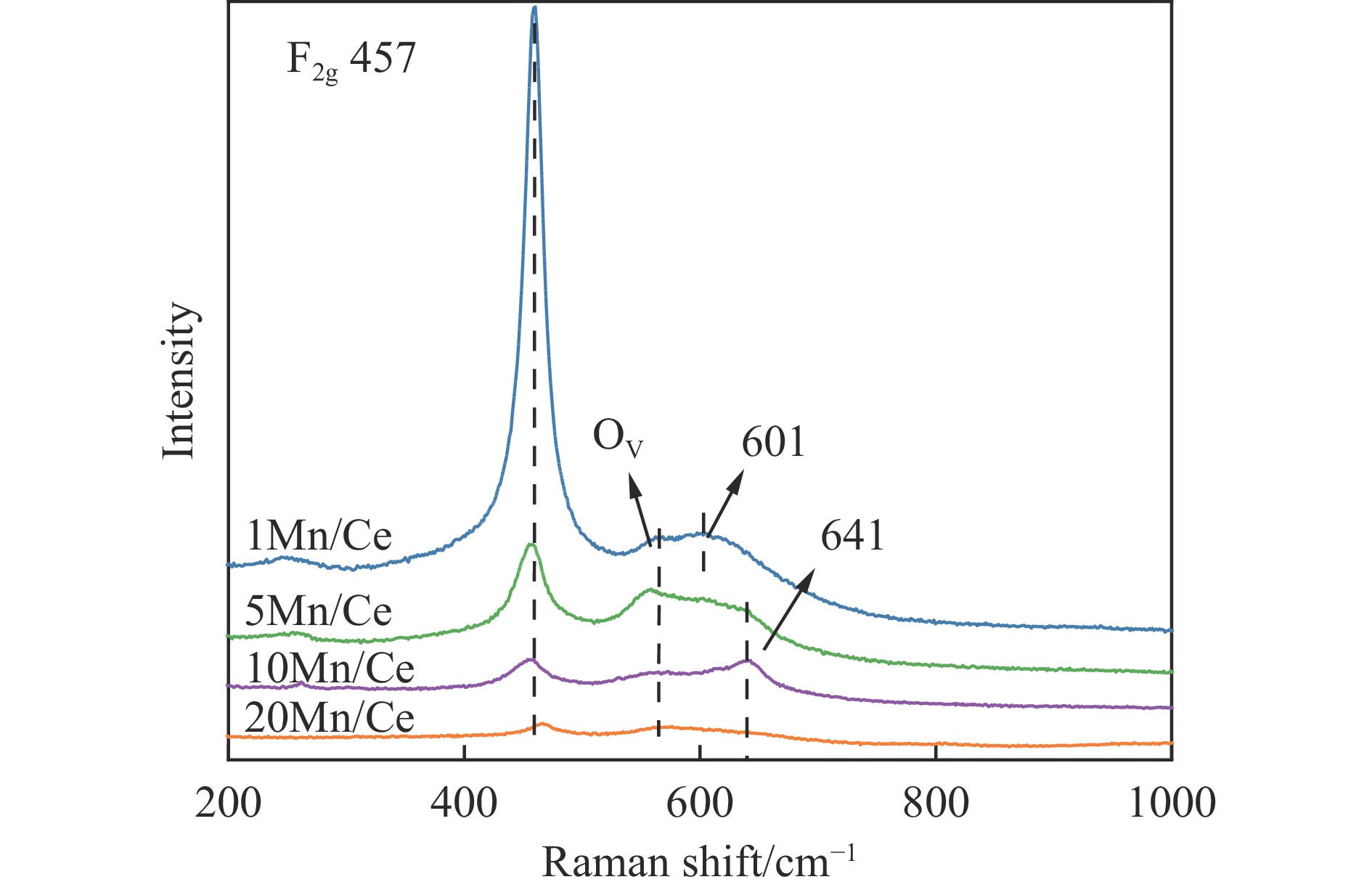
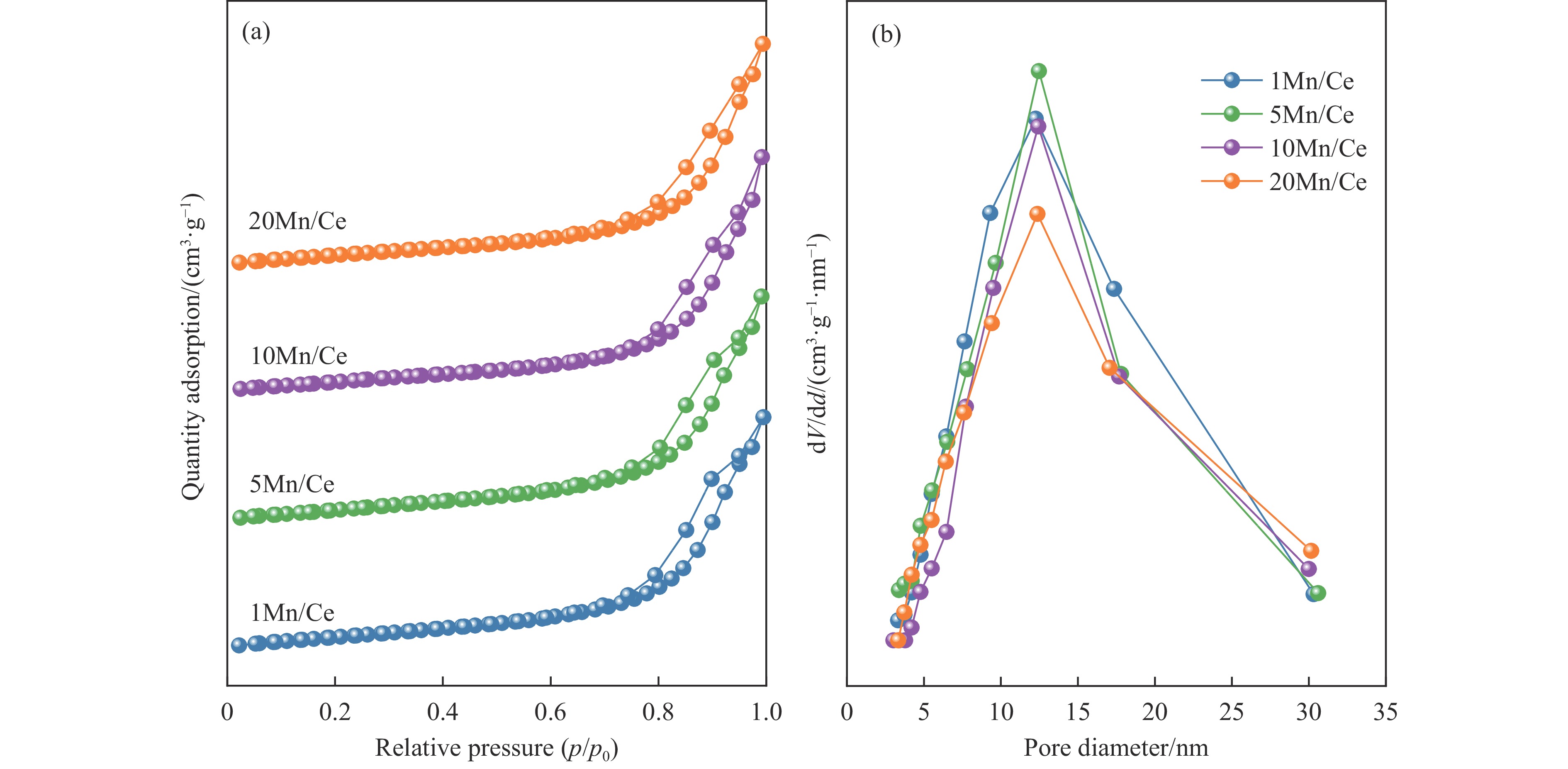
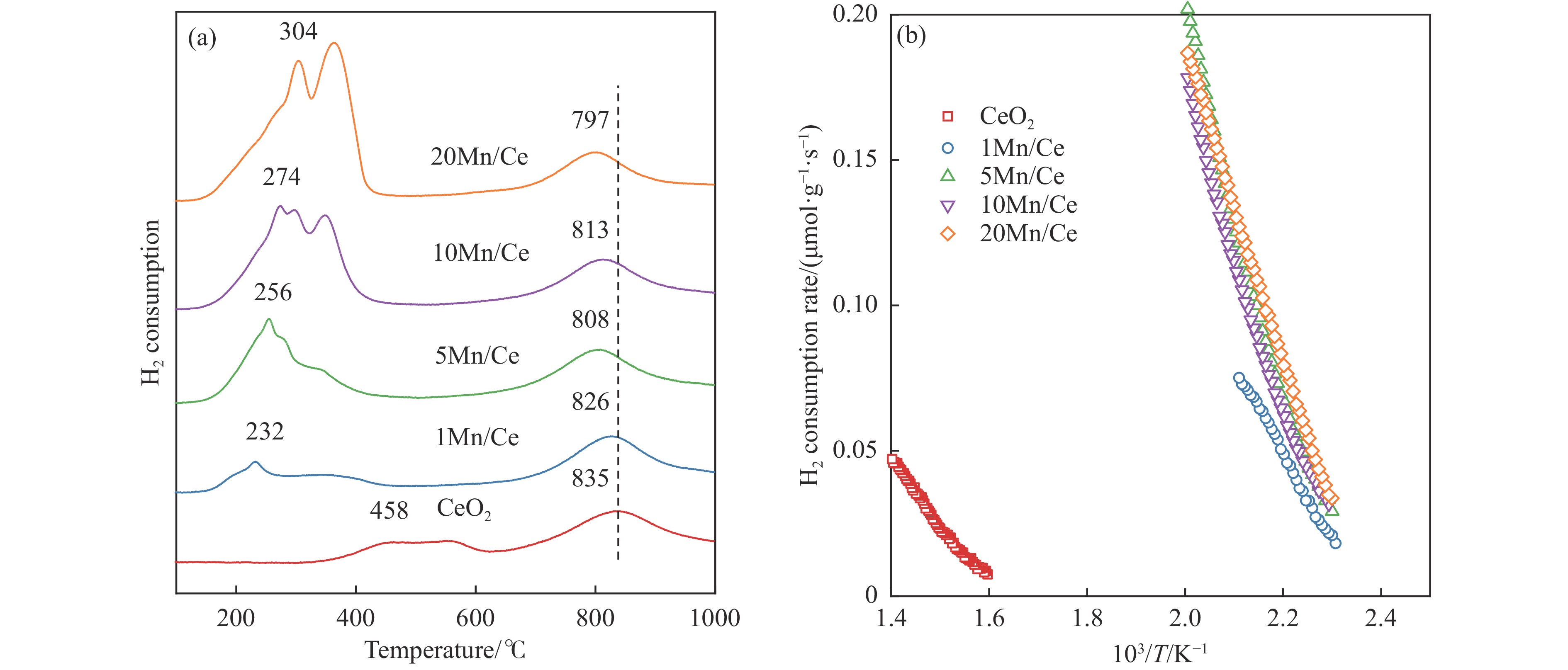
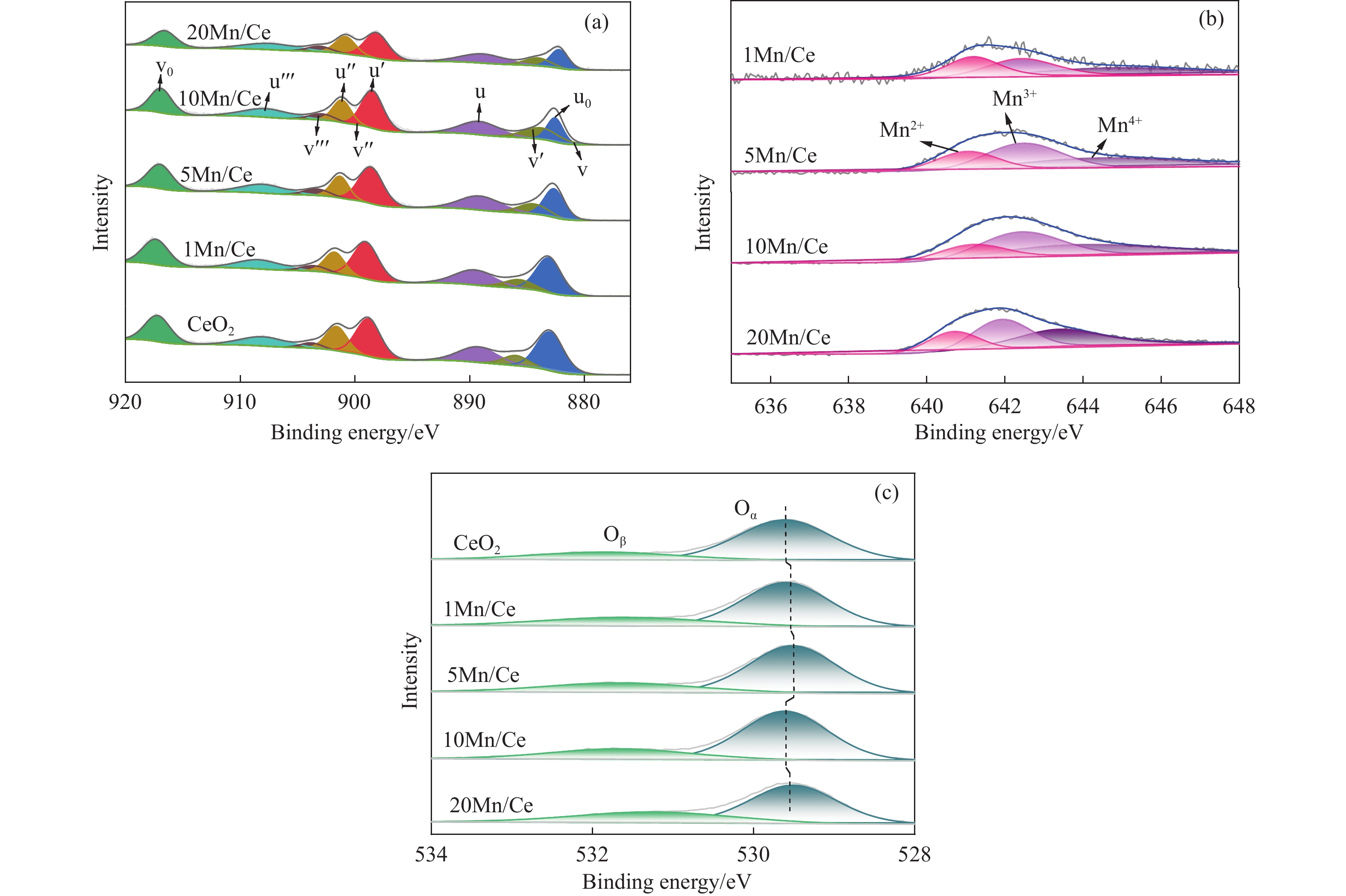
本研究,通过水热-浸渍两步法成功制备了不同Mn负载量的二元xMn/Ce(xMnOx/CeO2)催化剂,并评估了这些催化剂在甲苯催化氧化反应中的性能。研究结果表明,引入MnOx能显著提高催化剂的甲苯氧化活性。特别是当Mn负载量为10%(10Mn/Ce)时,在气体空速为60000 mL/(g·h)的条件下,t90(甲苯转化率达到90%时的温度)仅为233 ℃,显示出最优的甲苯催化氧化活性。这一结果说明,适量的MnOx加入能够显著提高催化剂的催化性能。通过X射线衍射(XRD)、拉曼光谱(Raman)、透射电子显微镜(TEM)、程序升温还原(H2-TPR)和X射线光电子能谱(XPS)等表征手段,发现MnOx的加入在MnOx与CeO2之间形成了界面效应,这显著改变了Mn/Ce催化剂的物理化学性质。由于界面效应的作用,不仅提高了10Mn/Ce催化剂中Ce3+、Mn3+离子的浓度以及氧空位的浓度,而且还降低了催化剂表面Ce−O键强度,使得表面晶格氧更易于参与甲苯的催化氧化,提升了催化剂的氧化还原性能,从而促进了甲苯的催化氧化。本研究不仅成功制备了具有优异甲苯氧化活性的Mn/Ce催化剂,而且揭示了其背后的界面效应机制,为VOCs高效氧化催化剂设计与制备提供了简单有效的方法与思路。
本研究,通过水热-浸渍两步法成功制备了不同Mn负载量的二元xMn/Ce(xMnOx/CeO2)催化剂,并评估了这些催化剂在甲苯催化氧化反应中的性能。研究结果表明,引入MnOx能显著提高催化剂的甲苯氧化活性。特别是当Mn负载量为10%(10Mn/Ce)时,在气体空速为60000 mL/(g·h)的条件下,t90(甲苯转化率达到90%时的温度)仅为233 ℃,显示出最优的甲苯催化氧化活性。这一结果说明,适量的MnOx加入能够显著提高催化剂的催化性能。通过X射线衍射(XRD)、拉曼光谱(Raman)、透射电子显微镜(TEM)、程序升温还原(H2-TPR)和X射线光电子能谱(XPS)等表征手段,发现MnOx的加入在MnOx与CeO2之间形成了界面效应,这显著改变了Mn/Ce催化剂的物理化学性质。由于界面效应的作用,不仅提高了10Mn/Ce催化剂中Ce3+、Mn3+离子的浓度以及氧空位的浓度,而且还降低了催化剂表面Ce−O键强度,使得表面晶格氧更易于参与甲苯的催化氧化,提升了催化剂的氧化还原性能,从而促进了甲苯的催化氧化。本研究不仅成功制备了具有优异甲苯氧化活性的Mn/Ce催化剂,而且揭示了其背后的界面效应机制,为VOCs高效氧化催化剂设计与制备提供了简单有效的方法与思路。

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024002
摘要:
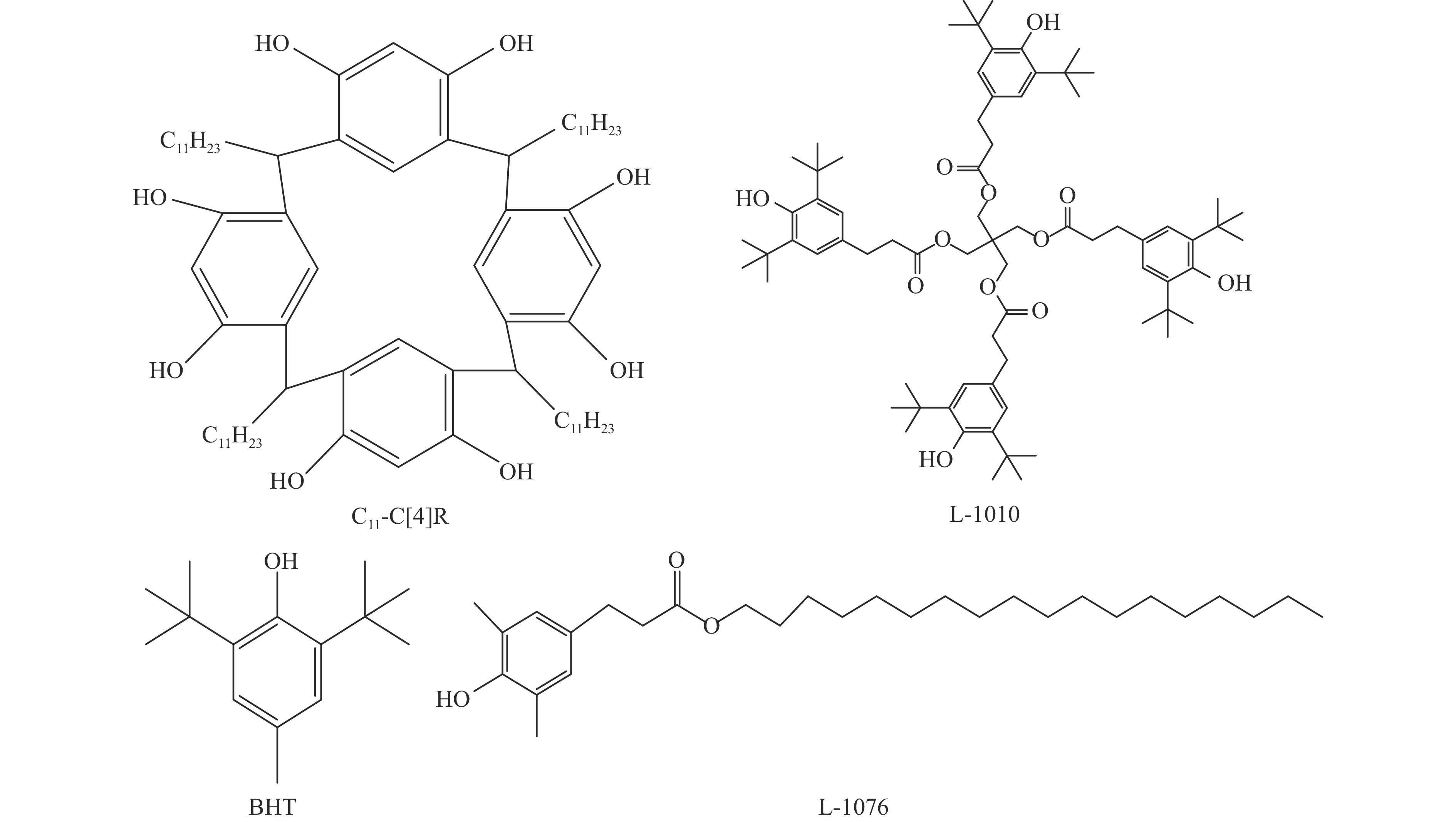
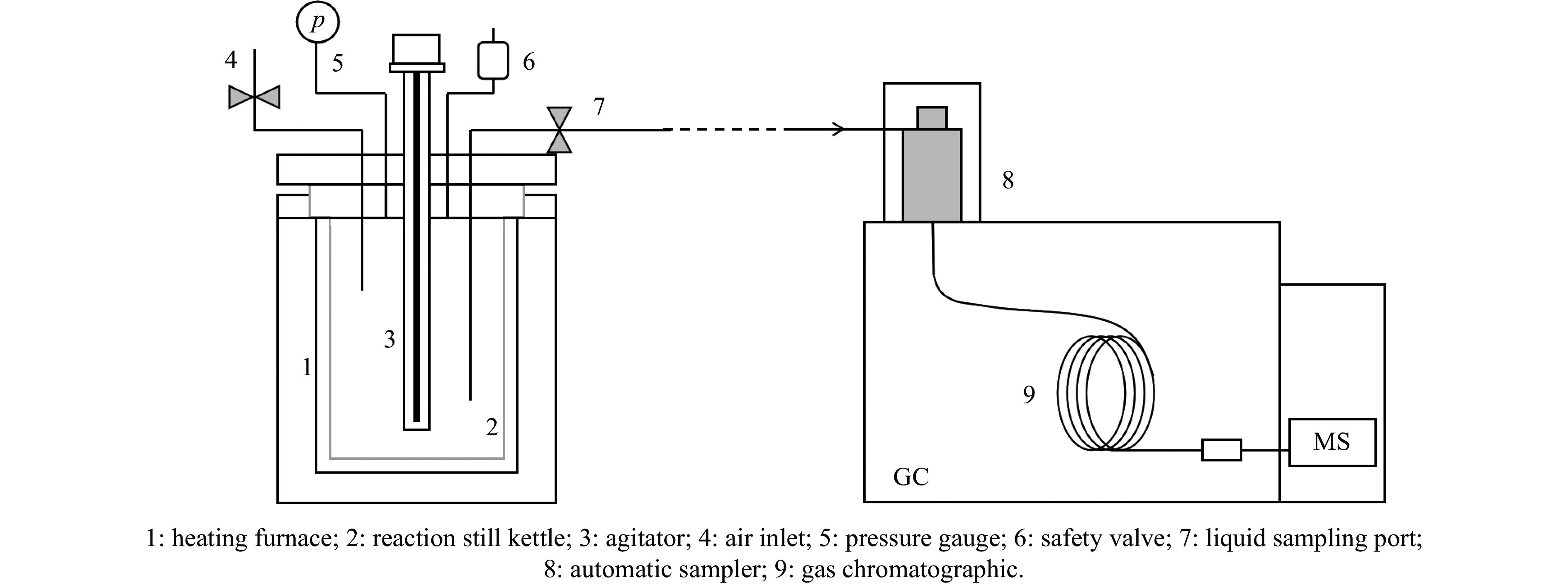
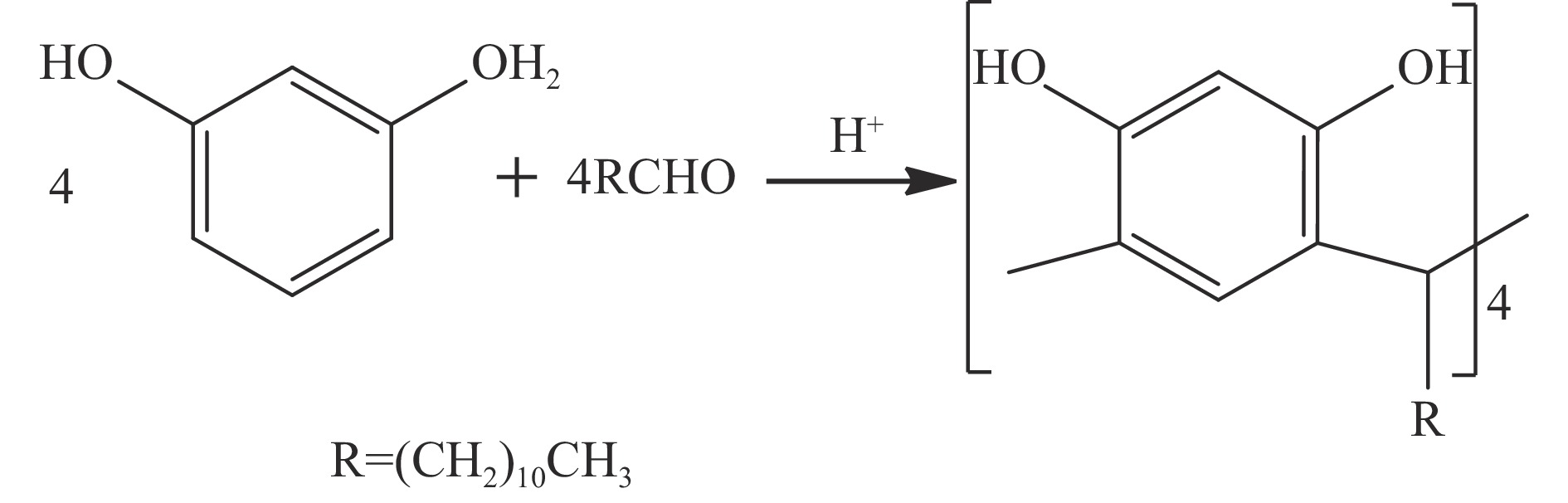
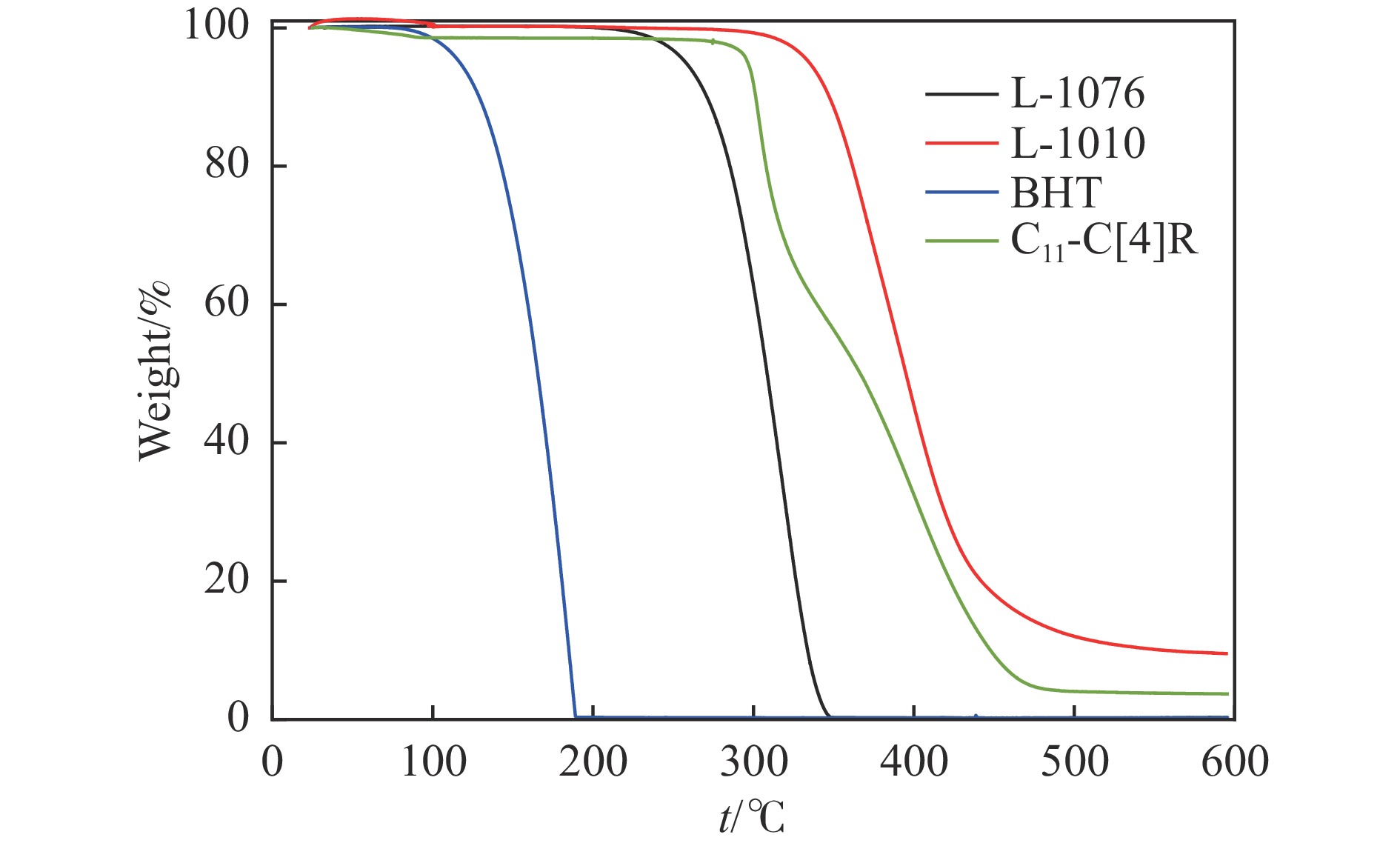
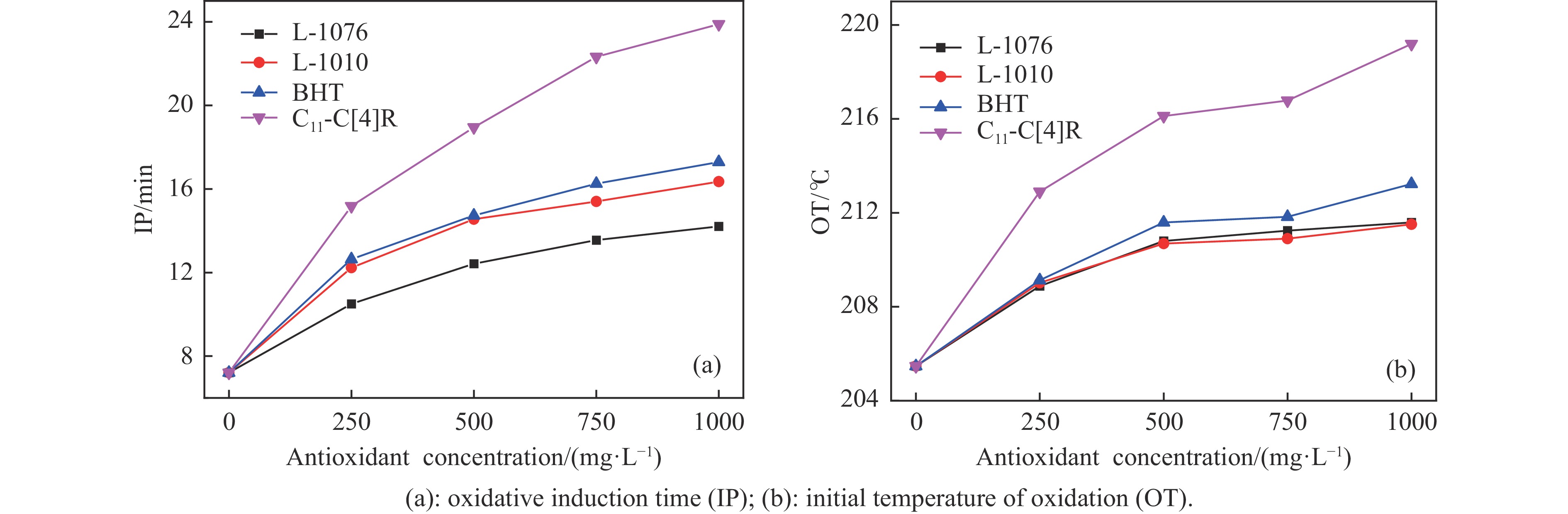
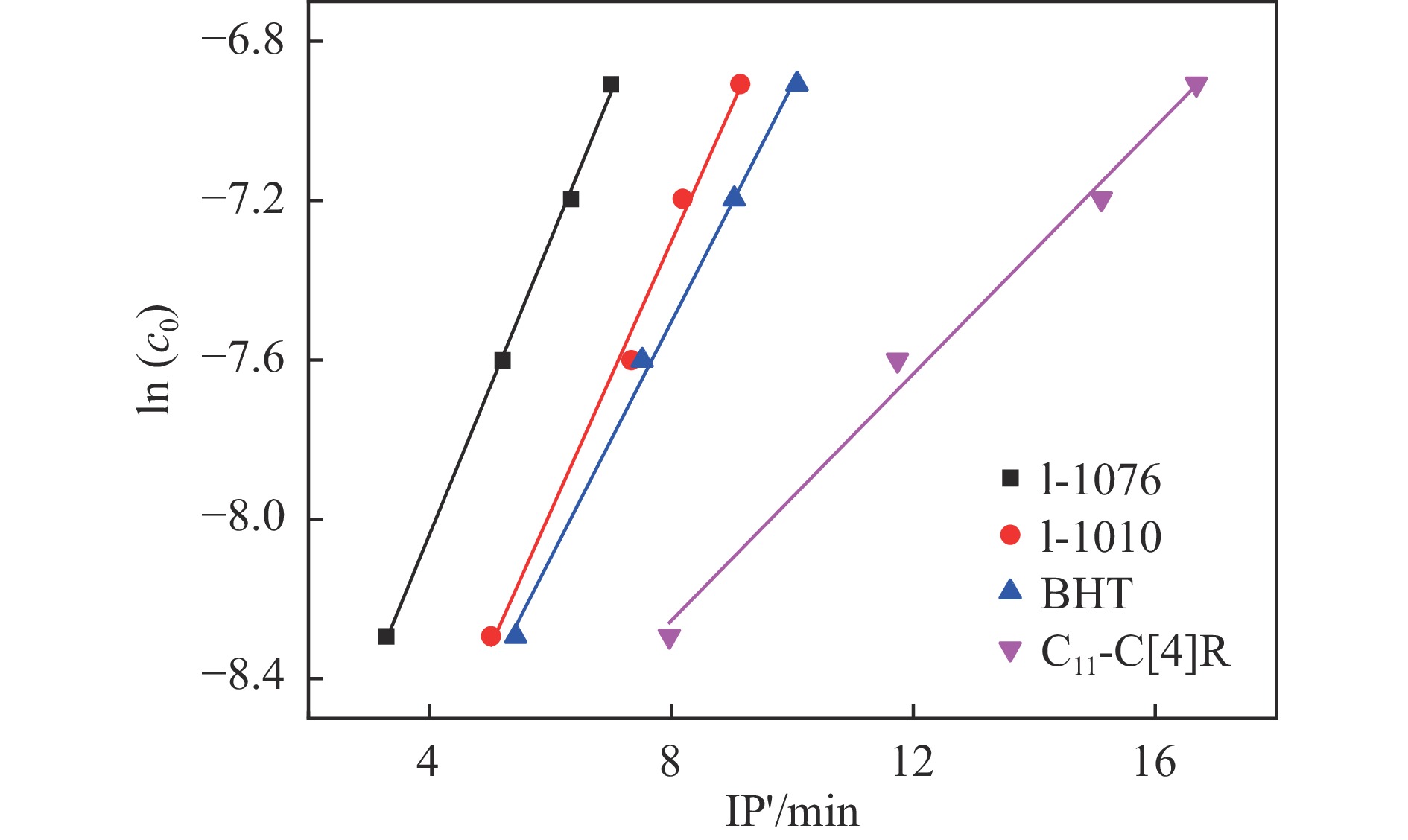
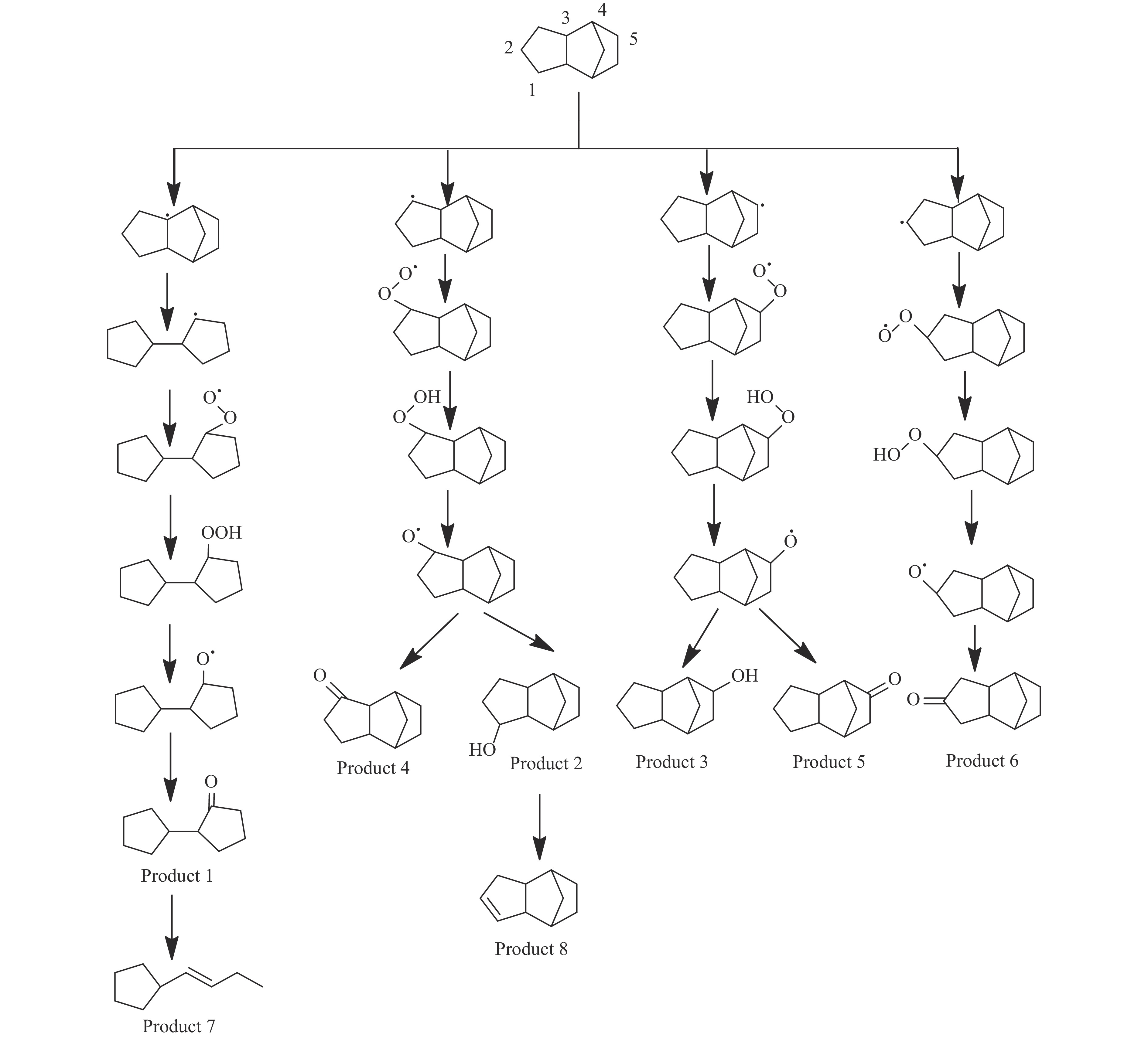
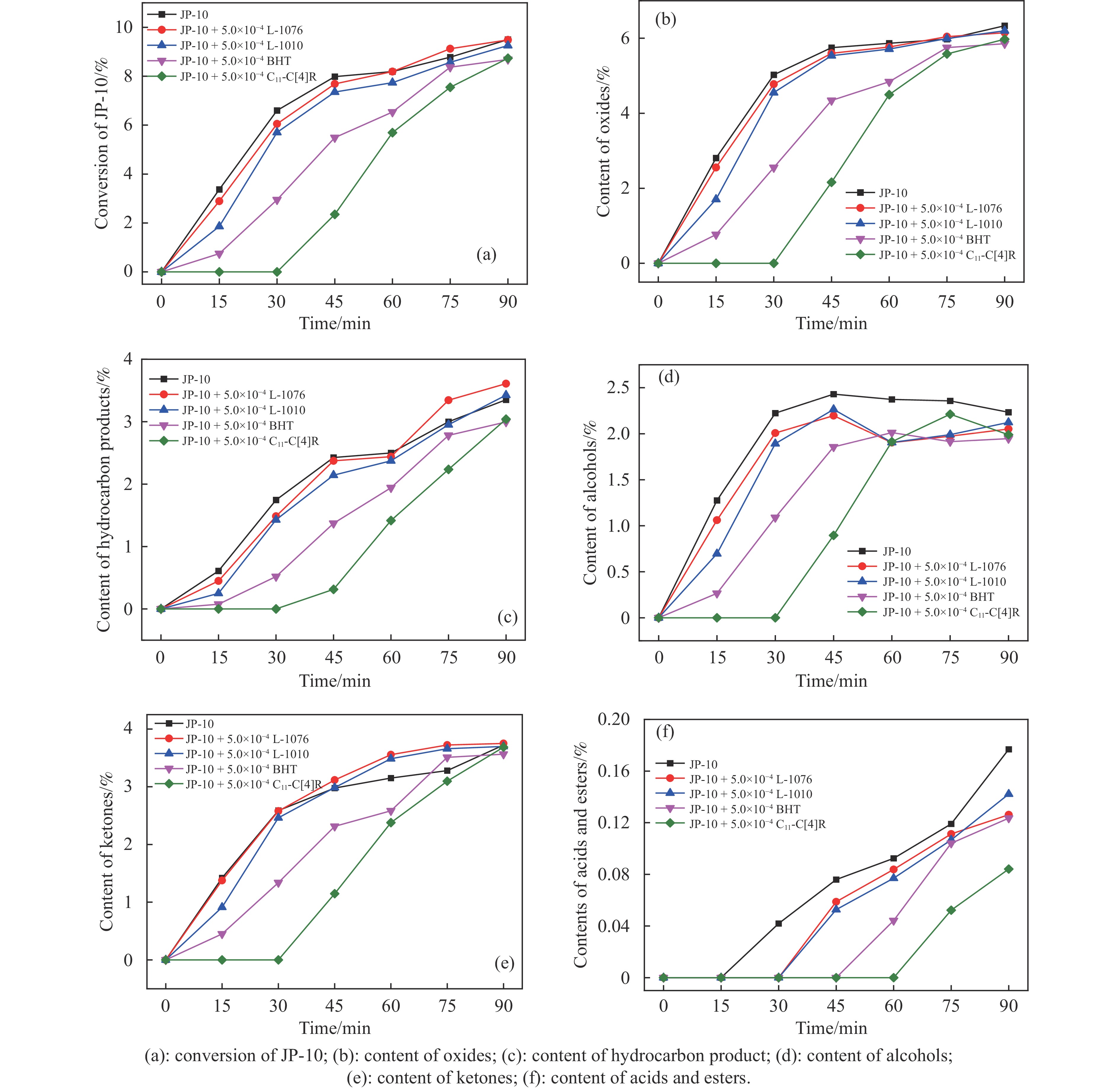
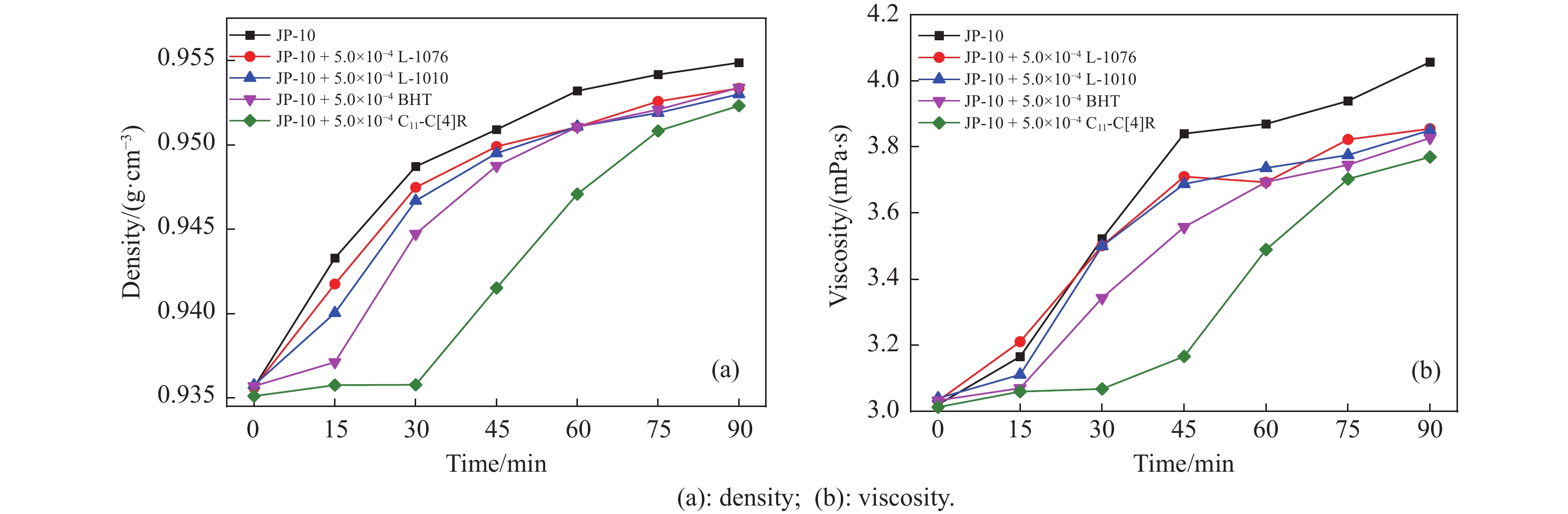
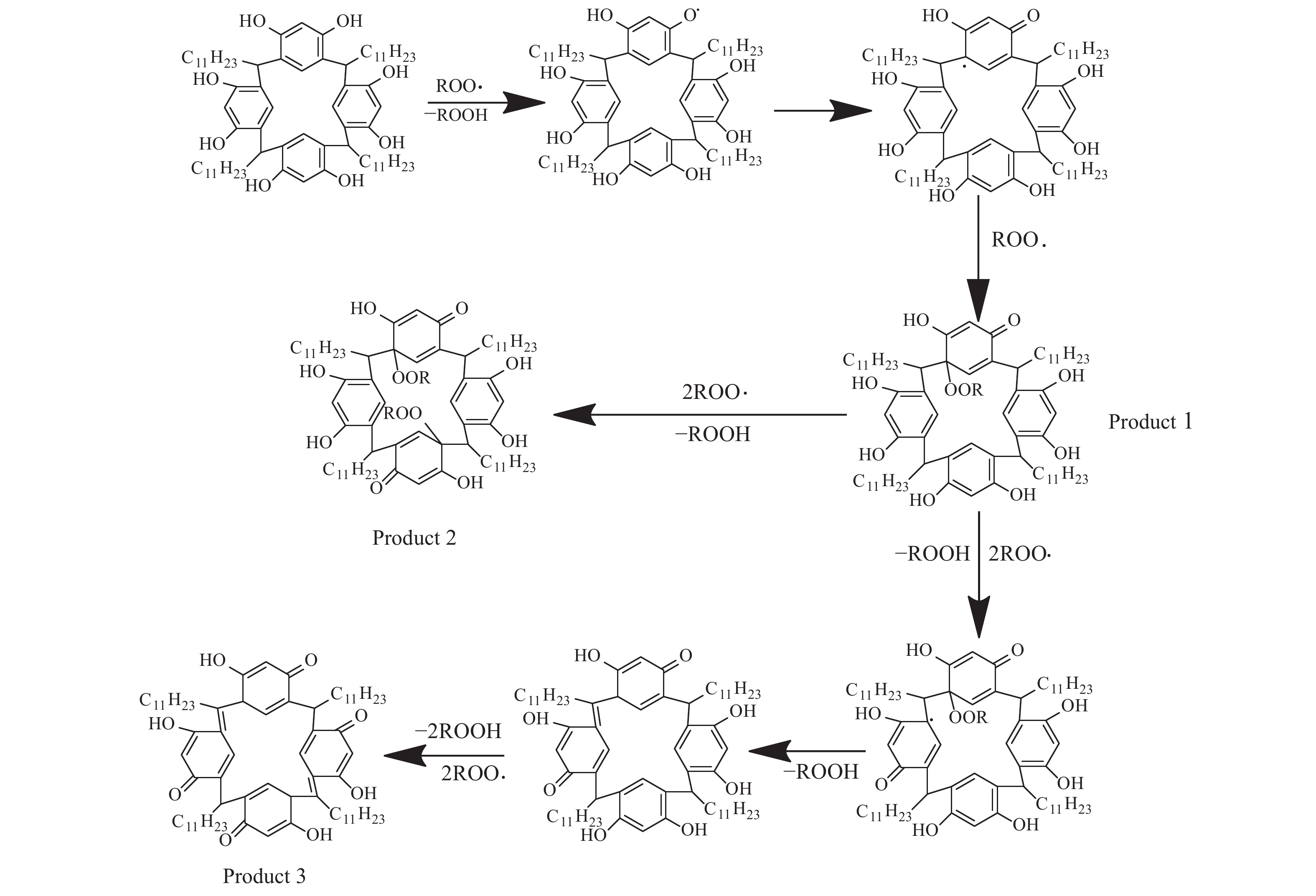
高密度吸热型碳氢燃料在热、催化等作用下会与溶解氧发生氧化反应,导致使用性能降低,进而威胁飞行安全。杯芳烃在与常用的酚类抗氧剂分子结构相似的前提下,拥有更好的热稳定性,可以在苛刻的工作环境中对燃料起到抗氧化作用。本实验合成了具有油溶性的C-十一烷基间苯二酚杯[4]芳烃(C11-C[4]R),考察其在高密度碳氢燃料JP-10中的抗氧化性能,并与商用抗氧剂2,6-二叔丁基-4-甲基苯酚(BHT)、四[β-(3,5-二叔丁基-4-羟基苯基)丙酸]季戊四醇酯(L-1010)和β-(3,5-二叔丁基-4-羟基苯基)丙酸十八碳醇酯(L-1076)等的抗氧化效果进行对比。高压差示扫描量热仪(PDSC)评价结果显示,四种抗氧化剂的效果排序为:C11-C [4] R > BHT > L-1010 > L-1076。还采用静态釜加速氧化法研究了JP-10的氧化反应历程,提出了C11-C[4]R在JP-10中的抗氧化机理。
高密度吸热型碳氢燃料在热、催化等作用下会与溶解氧发生氧化反应,导致使用性能降低,进而威胁飞行安全。杯芳烃在与常用的酚类抗氧剂分子结构相似的前提下,拥有更好的热稳定性,可以在苛刻的工作环境中对燃料起到抗氧化作用。本实验合成了具有油溶性的C-十一烷基间苯二酚杯[4]芳烃(C11-C[4]R),考察其在高密度碳氢燃料JP-10中的抗氧化性能,并与商用抗氧剂2,6-二叔丁基-4-甲基苯酚(BHT)、四[β-(3,5-二叔丁基-4-羟基苯基)丙酸]季戊四醇酯(L-1010)和β-(3,5-二叔丁基-4-羟基苯基)丙酸十八碳醇酯(L-1076)等的抗氧化效果进行对比。高压差示扫描量热仪(PDSC)评价结果显示,四种抗氧化剂的效果排序为:C11-C [4] R > BHT > L-1010 > L-1076。还采用静态釜加速氧化法研究了JP-10的氧化反应历程,提出了C11-C[4]R在JP-10中的抗氧化机理。

当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60408-6
摘要:
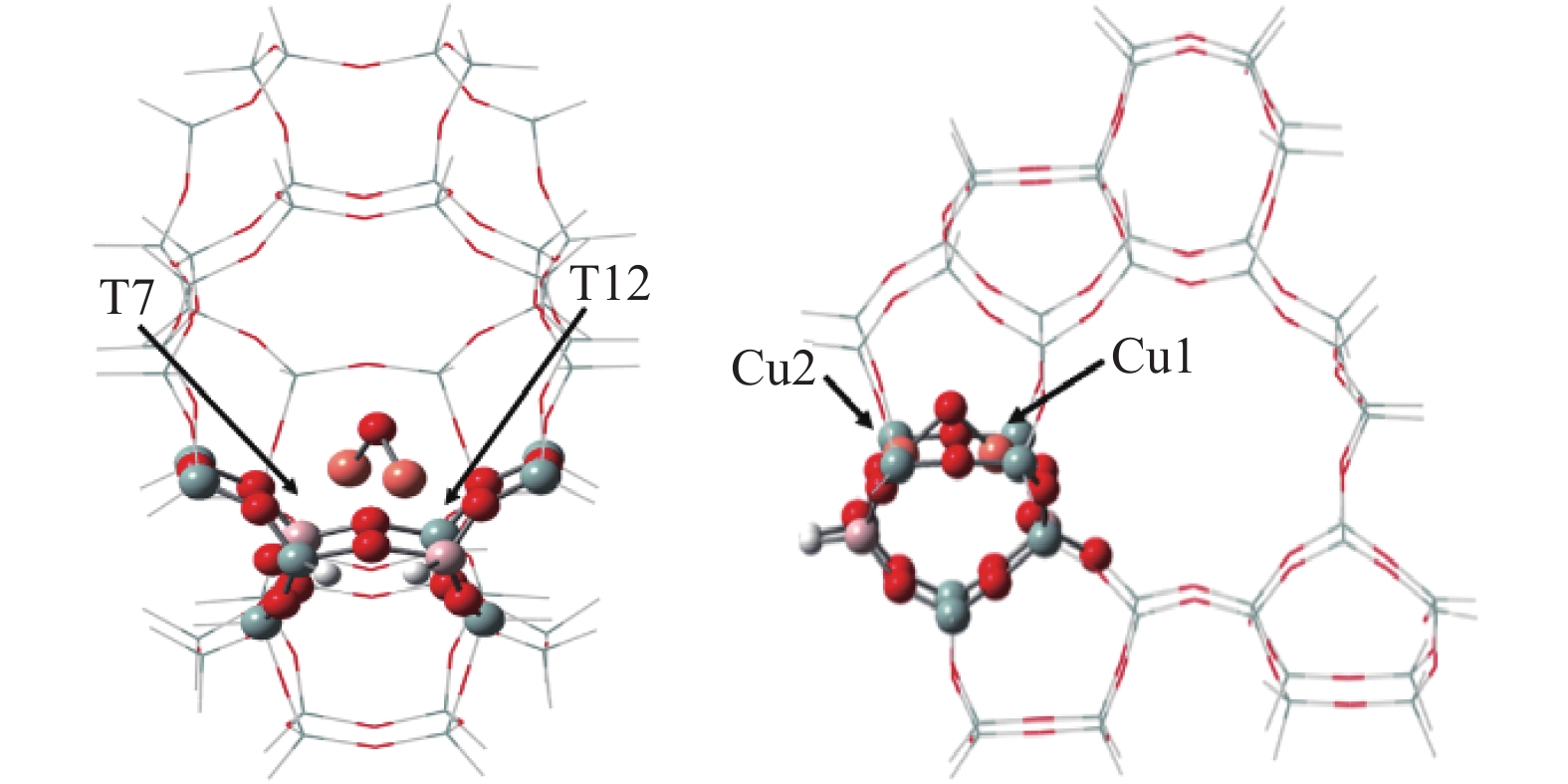
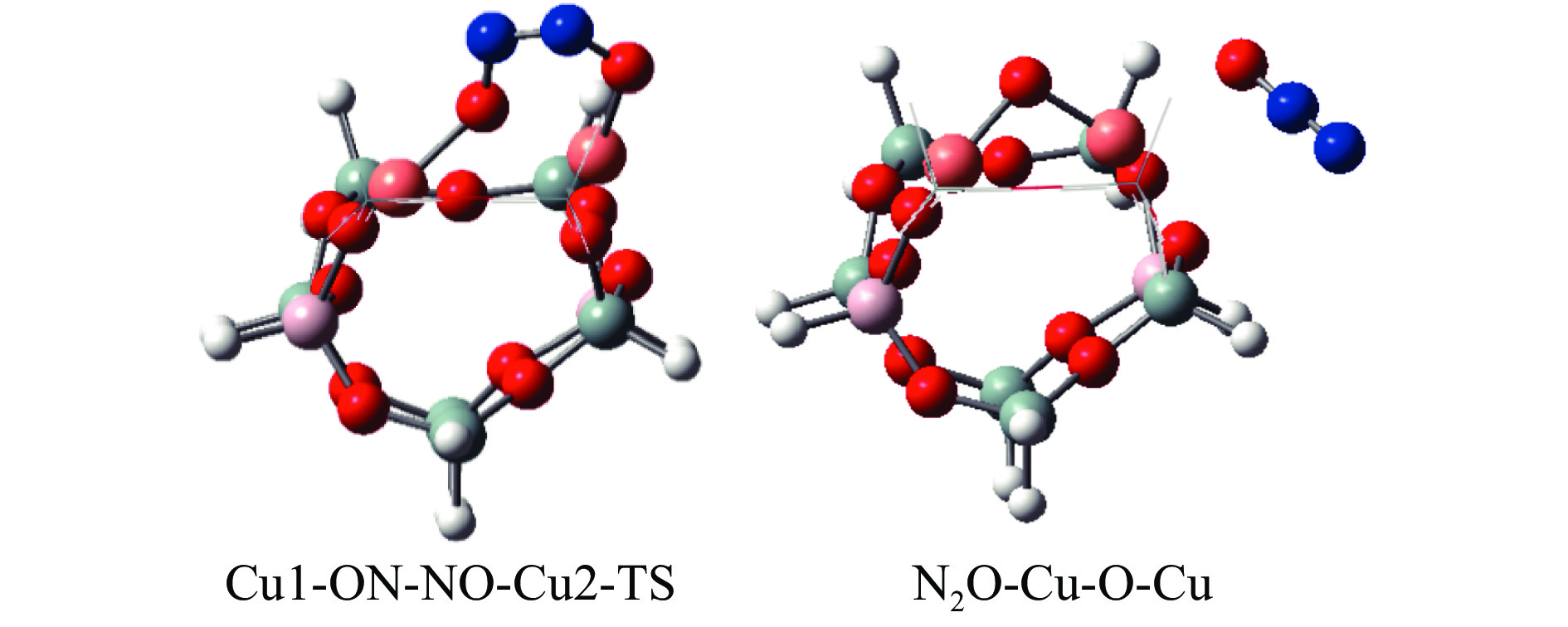
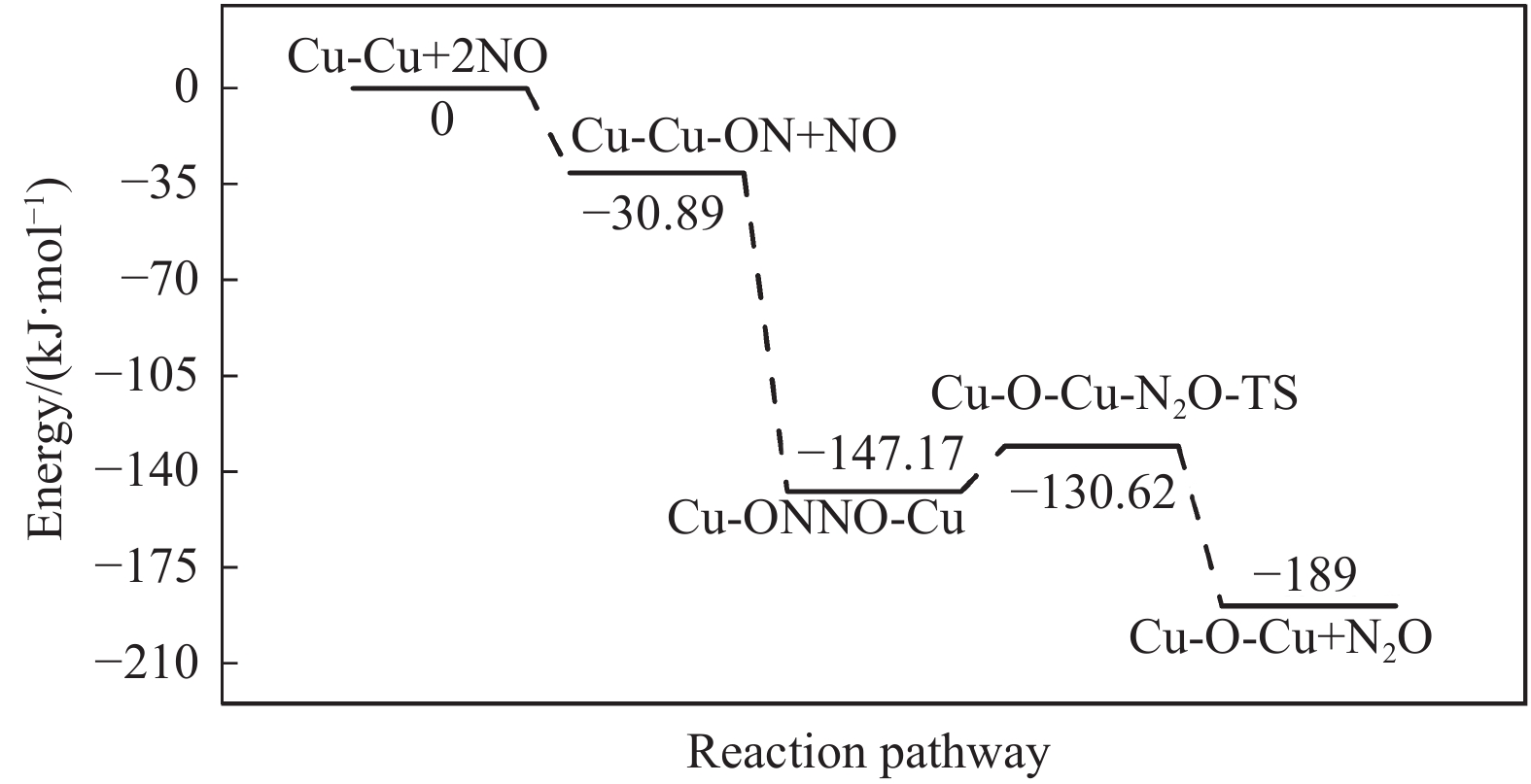
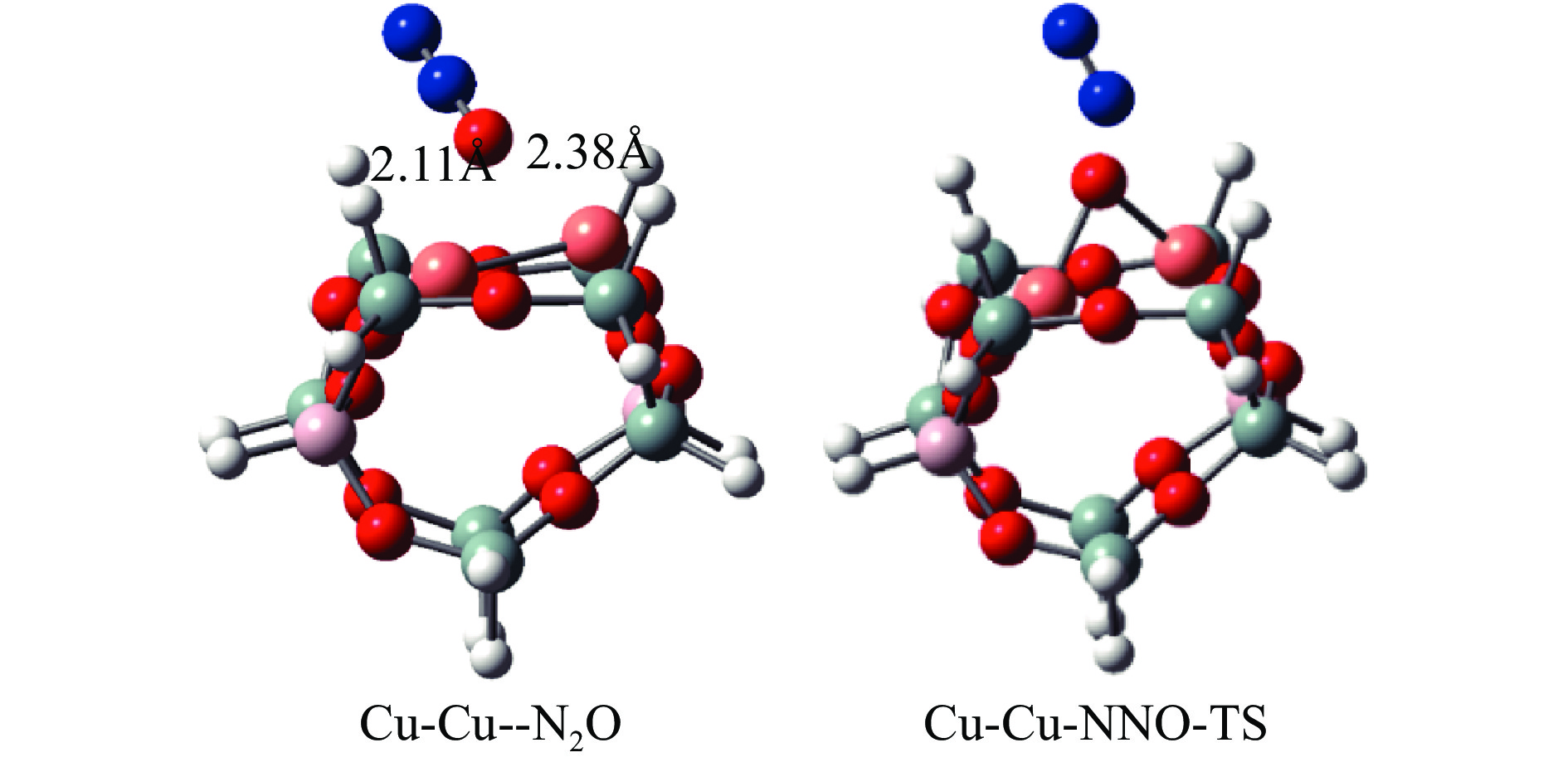
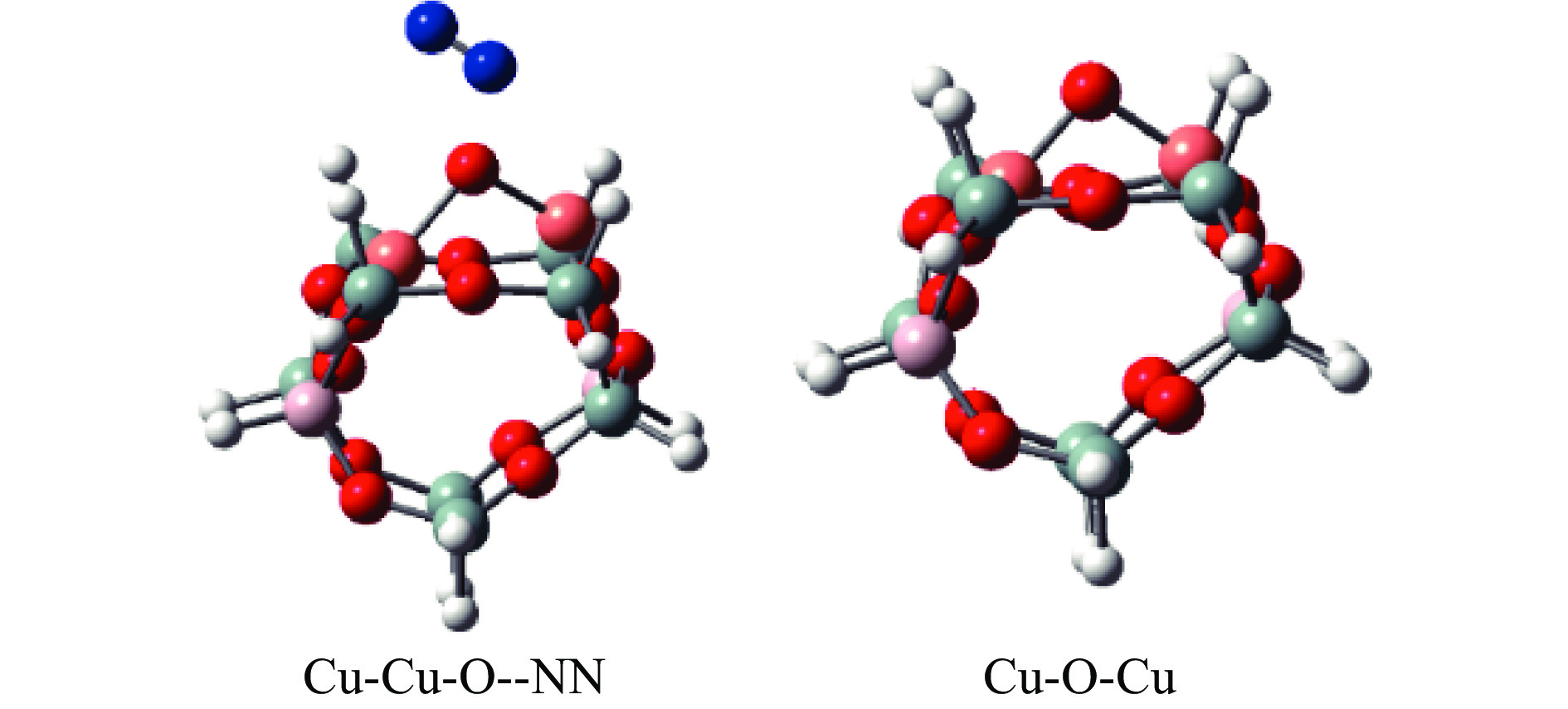
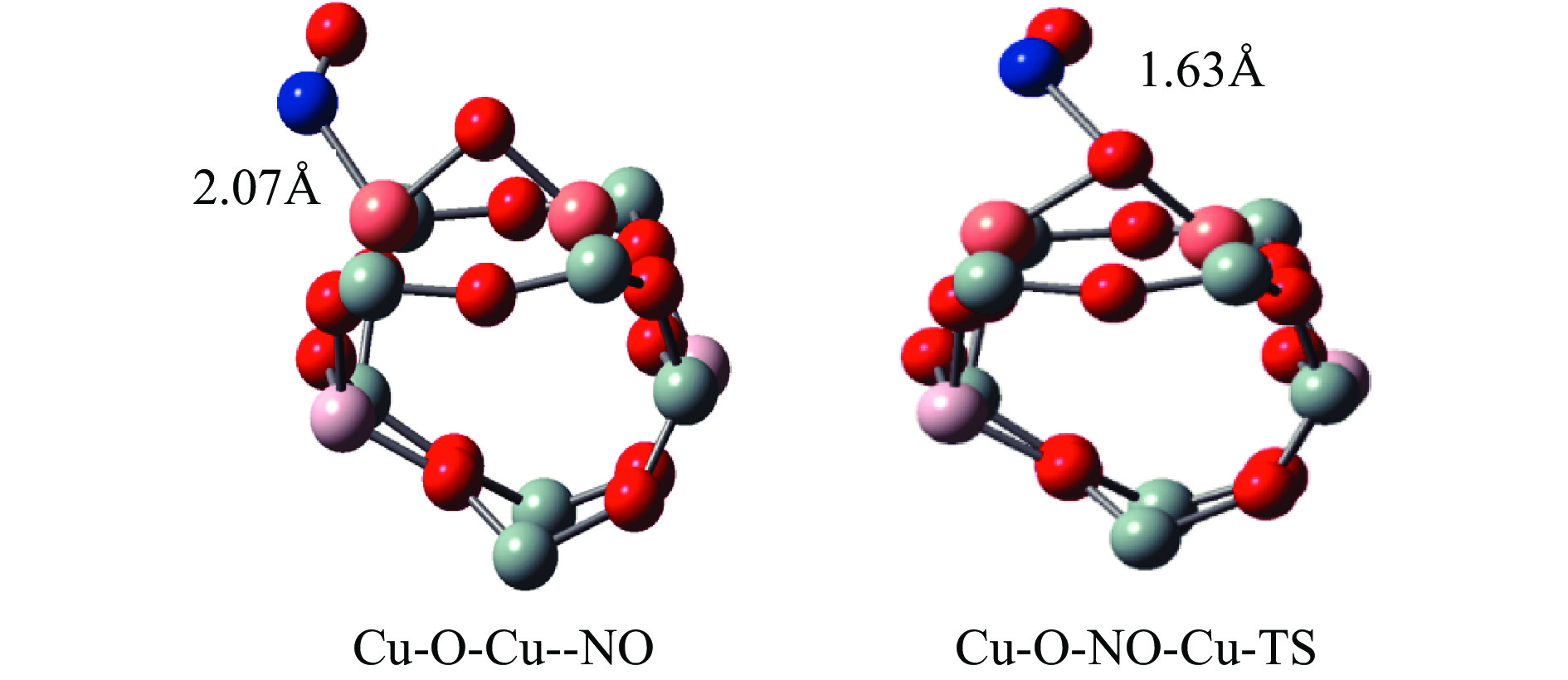
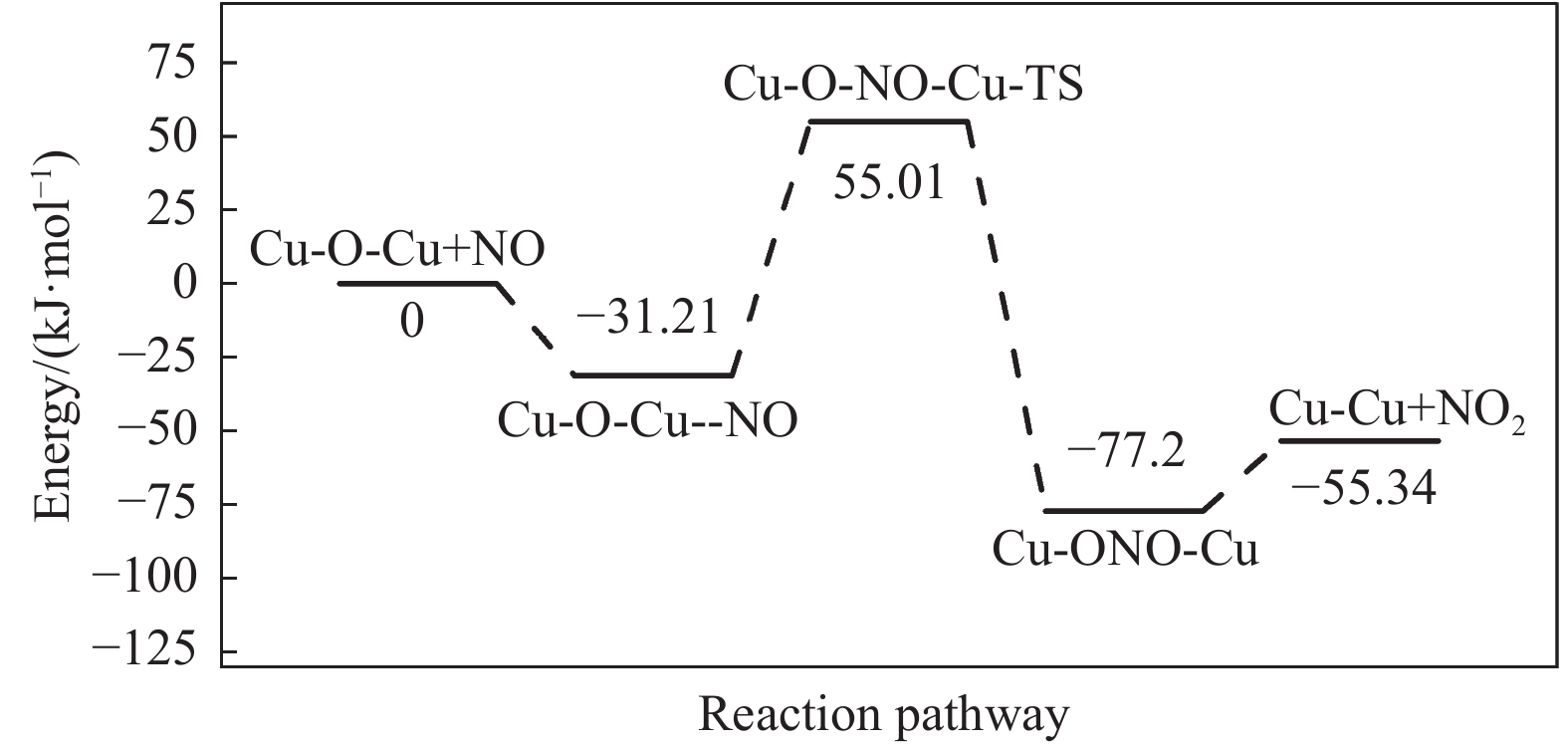
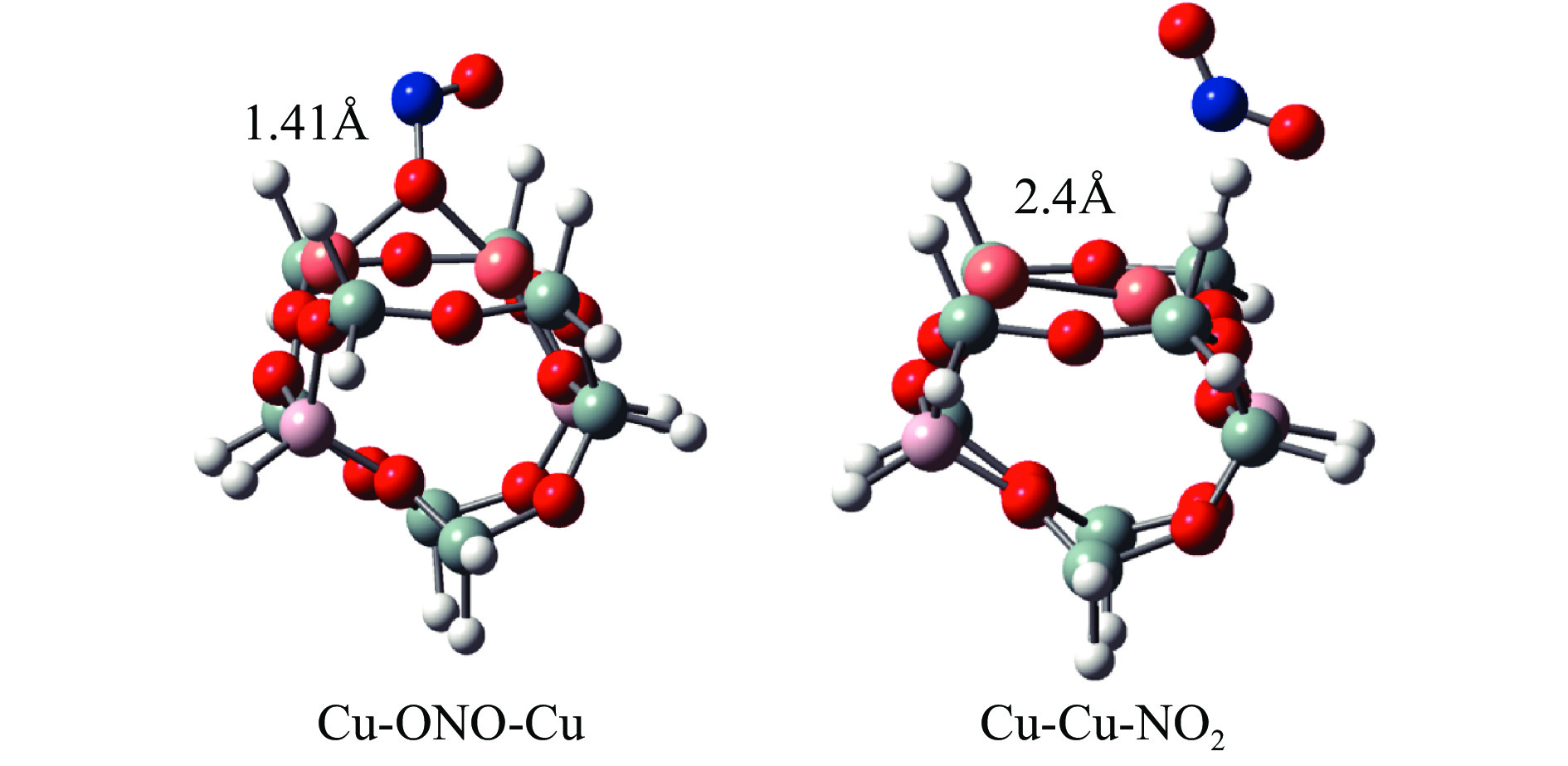
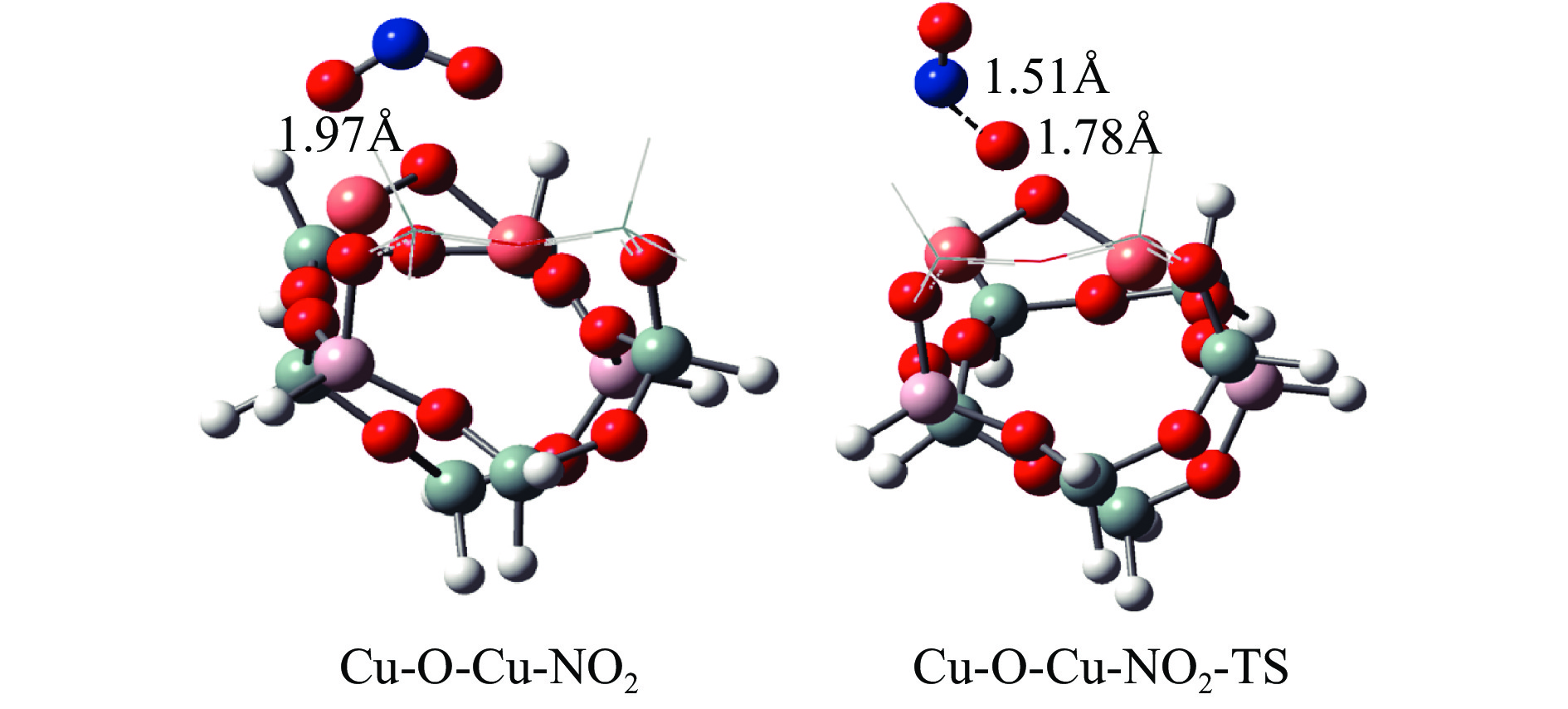
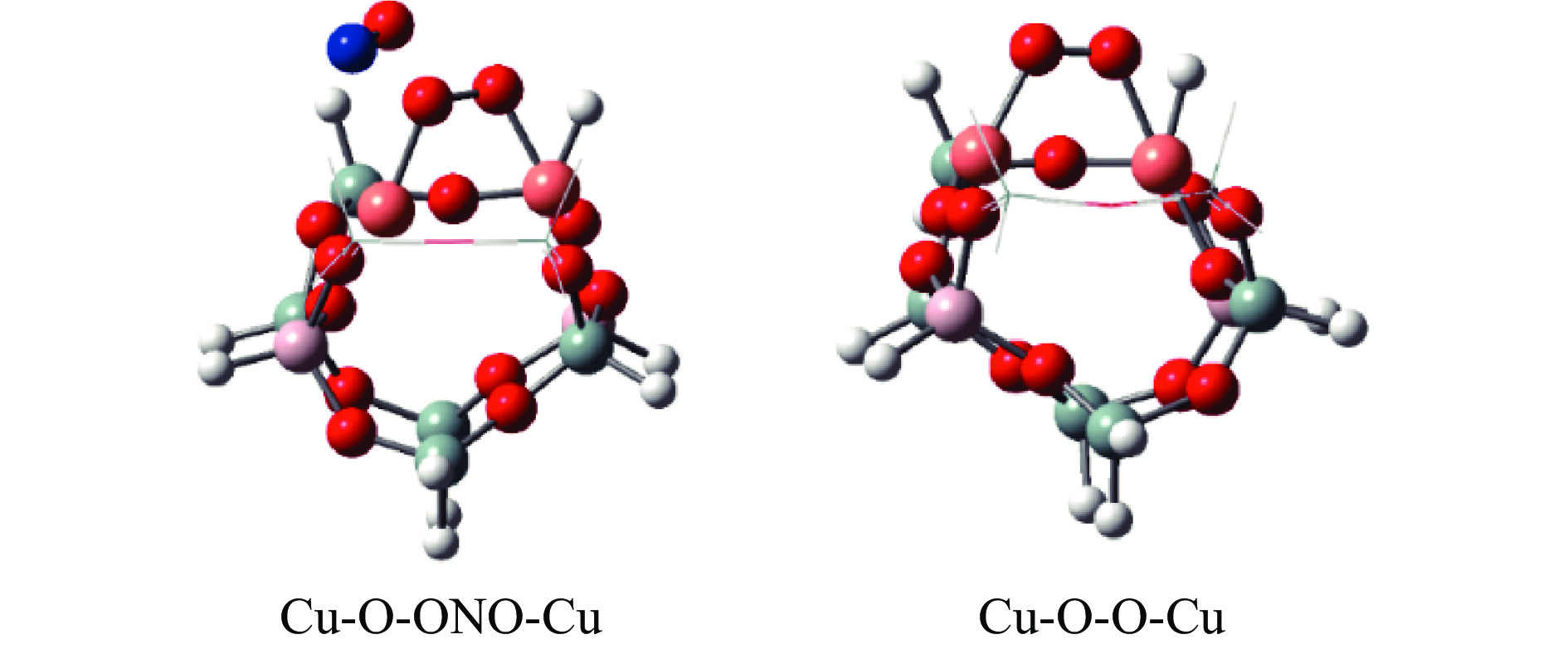
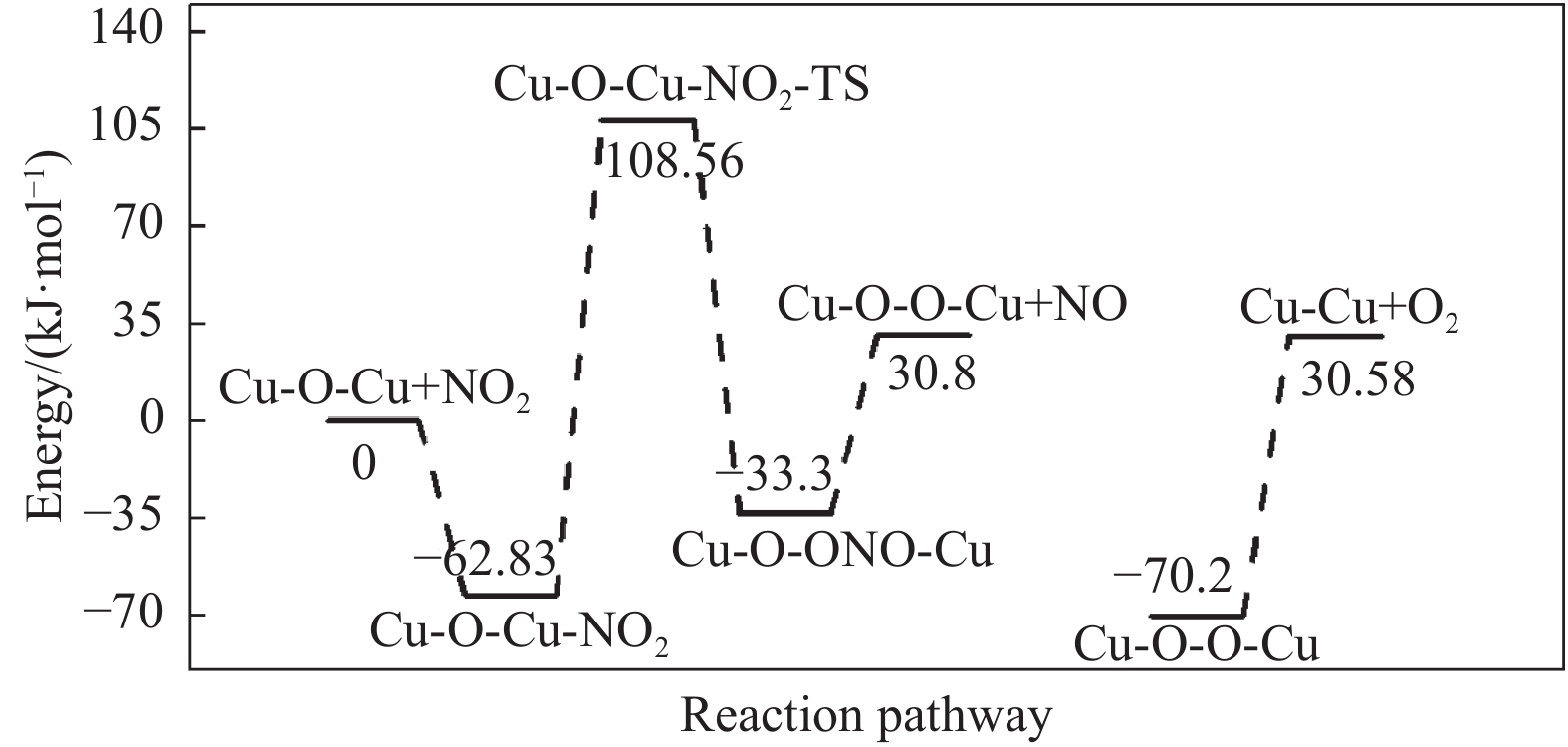
Cu-ZSM-5催化分解NO具有潜在的应用前景。为揭示NO在Cu-ZSM-5催化剂的催化分解机理,基于密度泛函模拟了NO在Cu-ZSM-5催化剂中短距离Cu+对上的吸附,并提出副产物N2O、NO2辅助催化分解NO的反应路径。计算结果表明,双核铜氧物种是Cu基催化剂的重要活性中心。催化分解NO过程中,副产物NO2在双核铜氧物种上的分解需要的活化能最高(为171.39 kJ/mol),N2O分解需要86.92 kJ/mol的活化能垒,表明NO2在活性位的分解难于N2O的分解。N2、O2的解析分别吸收28.43、100.78 kJ/mol的热量,限速步骤为O2的脱附。NO既作为反应物,同时又是催化过程中Cu-ZSM-5催化剂活性中心实现氧化还原循环的关键还原剂。
Cu-ZSM-5催化分解NO具有潜在的应用前景。为揭示NO在Cu-ZSM-5催化剂的催化分解机理,基于密度泛函模拟了NO在Cu-ZSM-5催化剂中短距离Cu+对上的吸附,并提出副产物N2O、NO2辅助催化分解NO的反应路径。计算结果表明,双核铜氧物种是Cu基催化剂的重要活性中心。催化分解NO过程中,副产物NO2在双核铜氧物种上的分解需要的活化能最高(为171.39 kJ/mol),N2O分解需要86.92 kJ/mol的活化能垒,表明NO2在活性位的分解难于N2O的分解。N2、O2的解析分别吸收28.43、100.78 kJ/mol的热量,限速步骤为O2的脱附。NO既作为反应物,同时又是催化过程中Cu-ZSM-5催化剂活性中心实现氧化还原循环的关键还原剂。

当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60410-4
摘要:
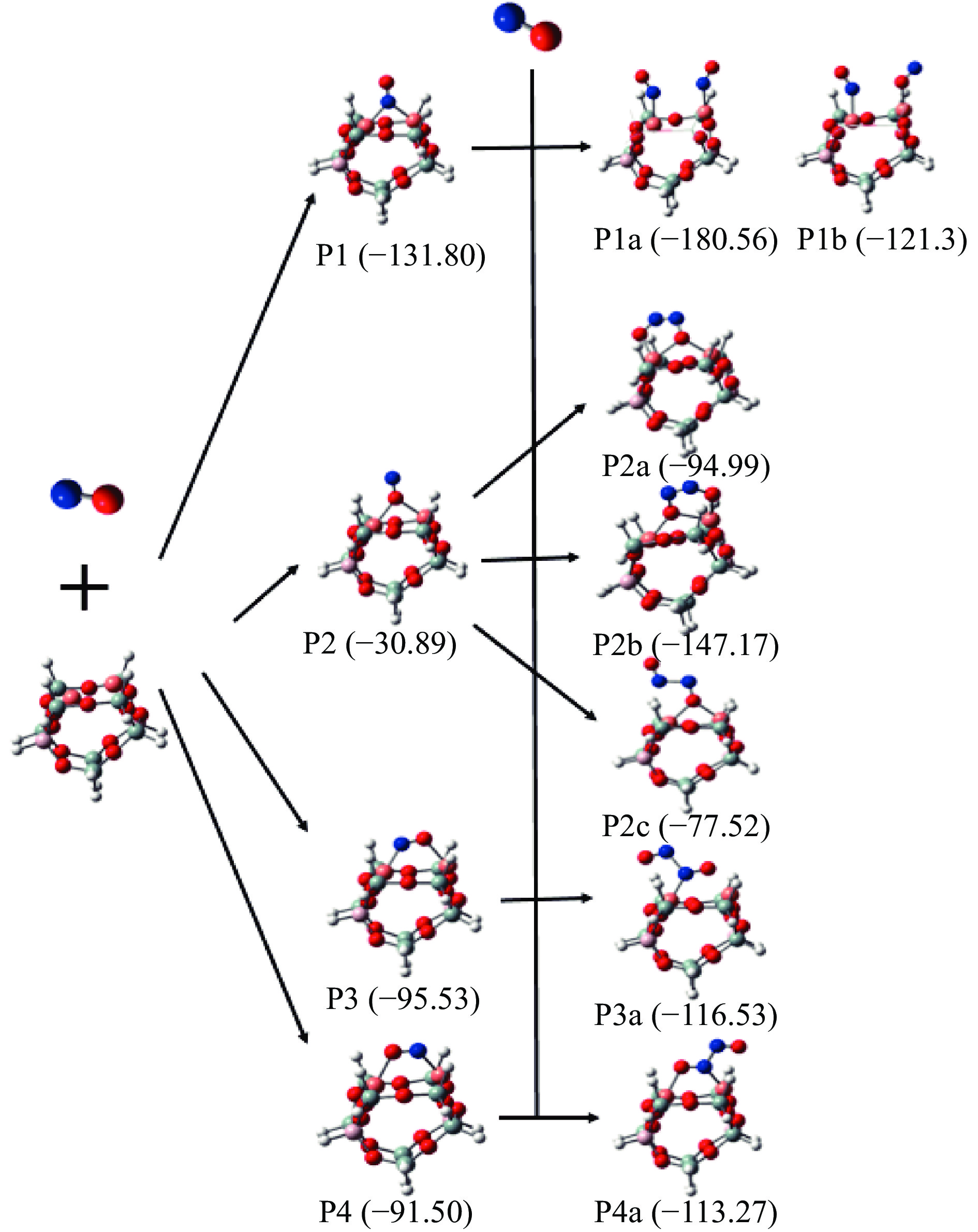
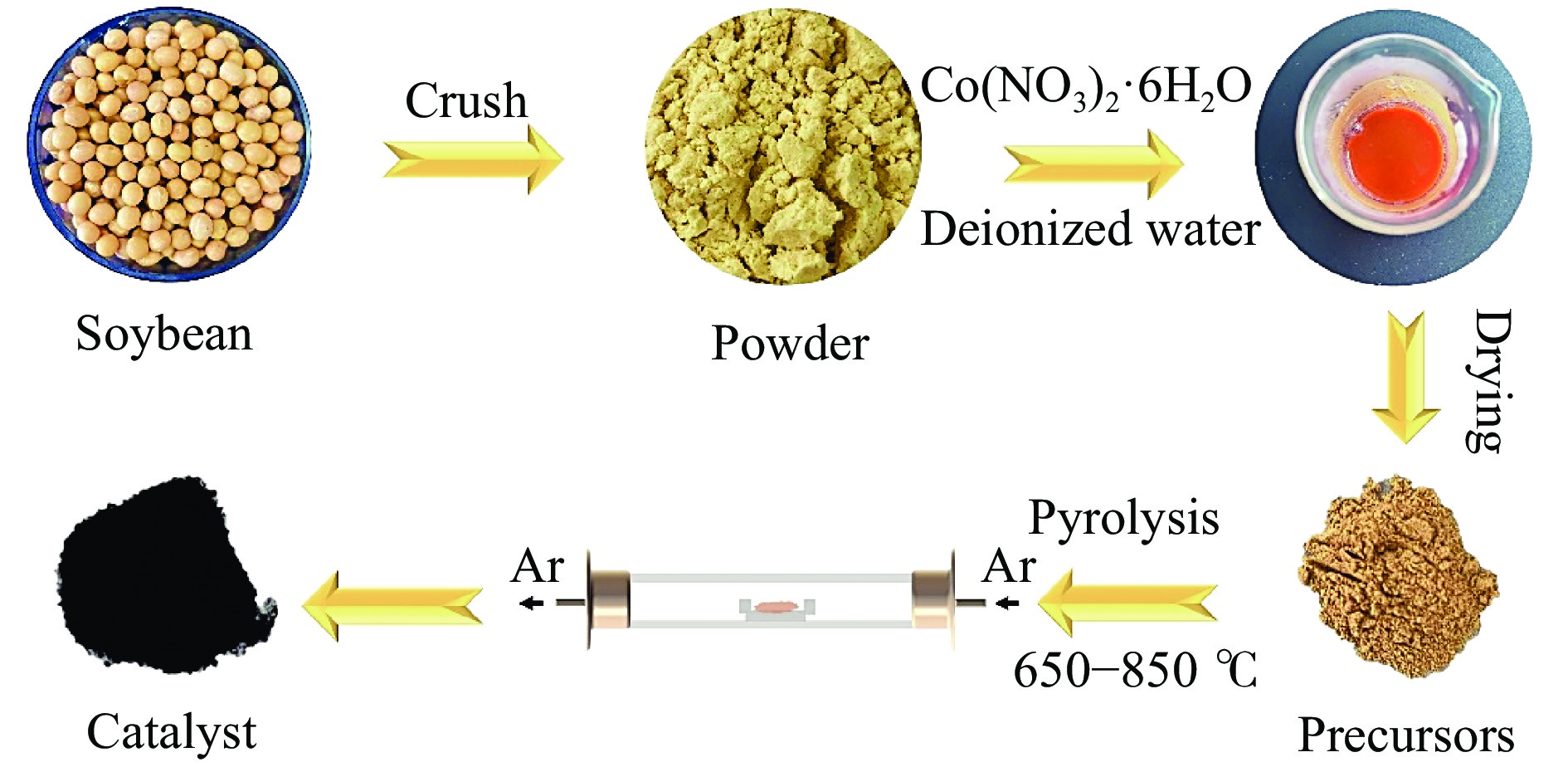
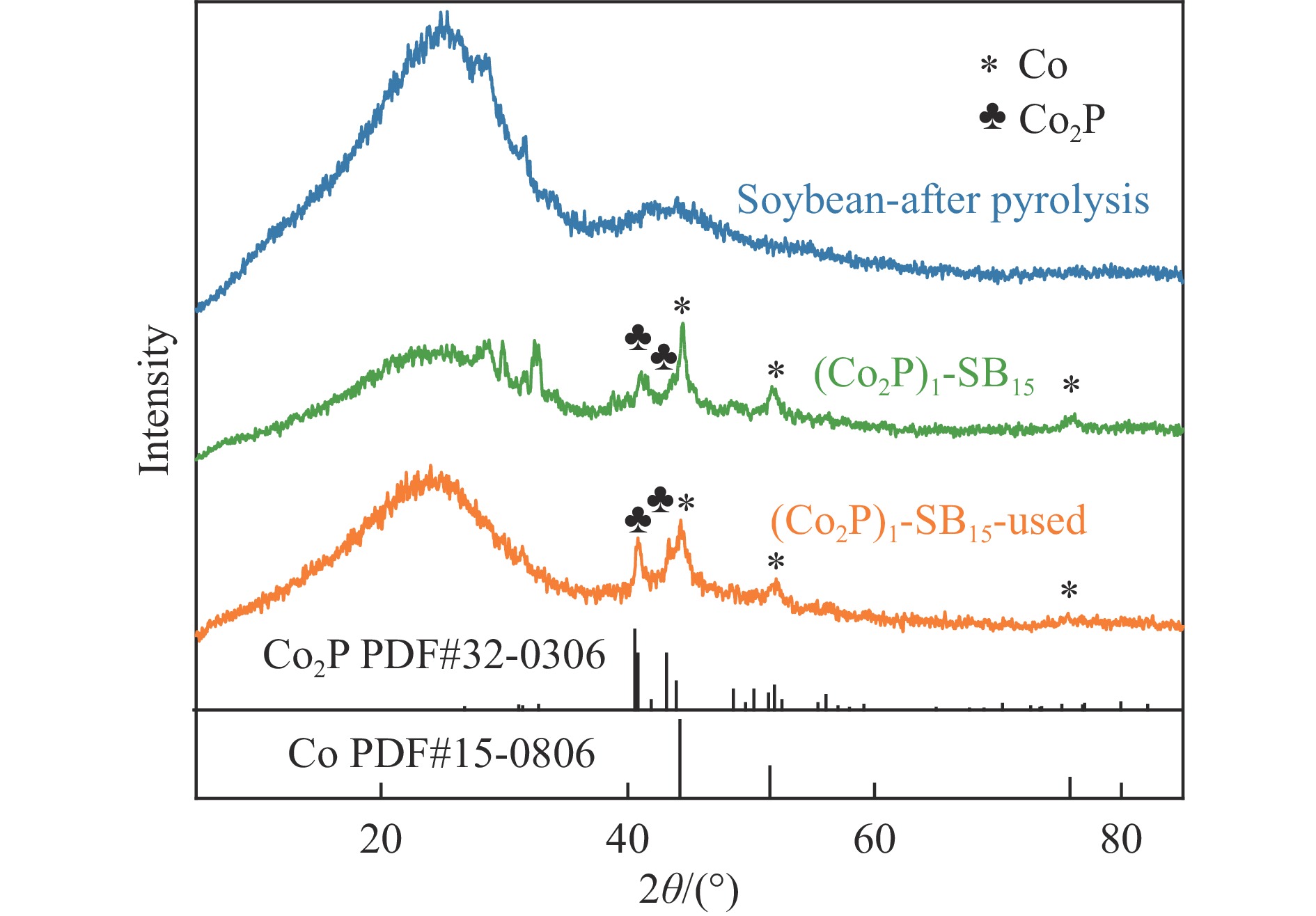
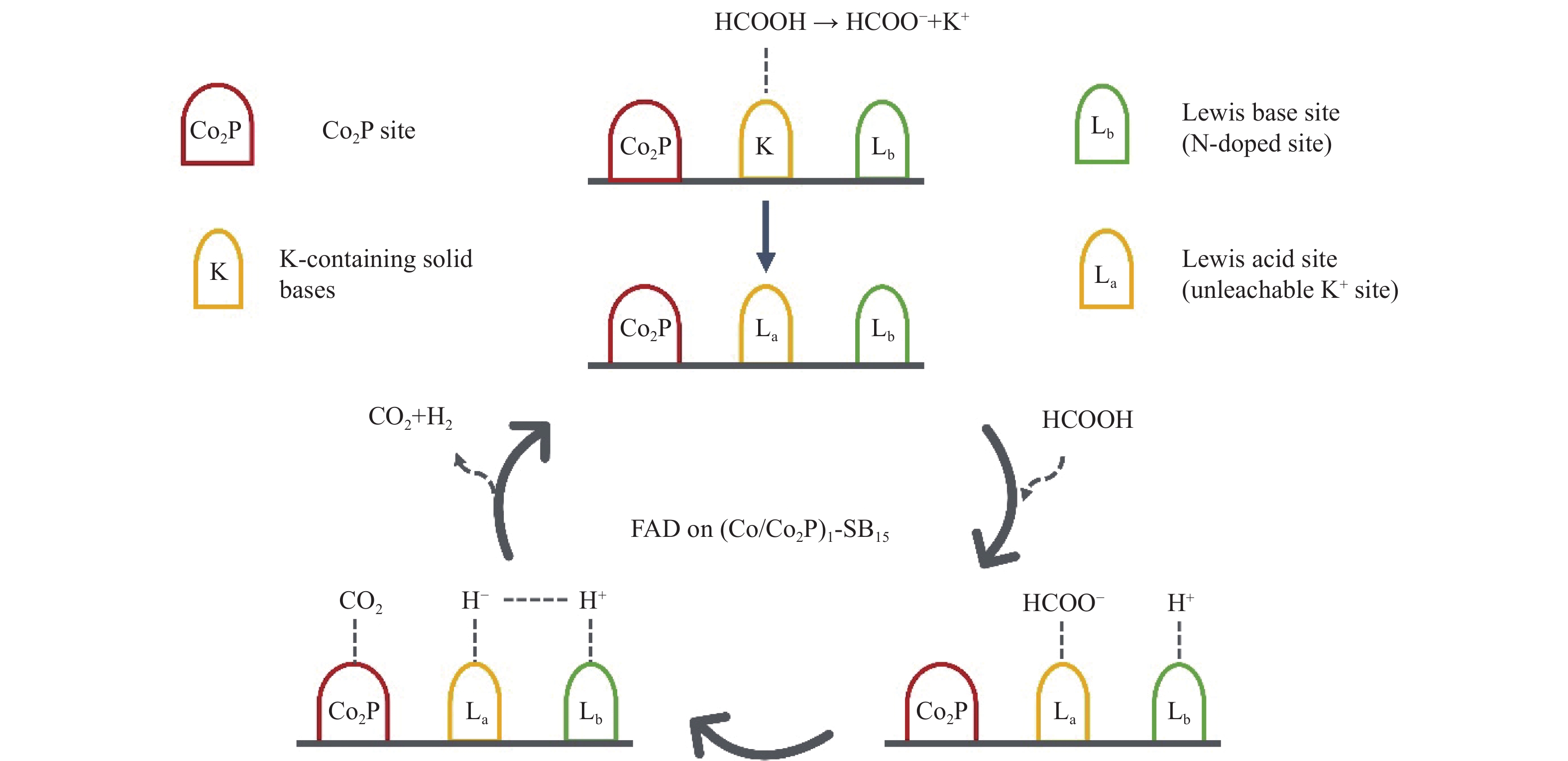
Formic acid (FA) is a sustainable liquid organic hydrogen carrier and the catalyst for hydrogen production from FA has received significant attention. However, the development of efficient non-noble metal catalysts still remains challenges. In this work, we provide a technologically rather simple and environmental-friendly strategy to synthesize Co2P catalyst for dehydrogenation of FA by pyrolyzing soybean powder and cobalt salt. The K-containing solid bases in catalyst could act as Lewis acid sites for the HCOO− intermediate adsorption while the self-doped N could act as Lewis base sites to enhance the H+ adsorption. The P contained in soybean could combine with Co to form Co2P for H−C bond cleavage of HCOO−. At a Co(NO3)2·6H2O/soybean mass ratio of 1∶15, the as prepared Co2P catalyst demonstrated a gas production rate of 237.47 mL/(g·h) and a good stability. This study provides a novel strategy to develop non-noble metal heterogeneous catalysts for FA dehydrogenation.
Formic acid (FA) is a sustainable liquid organic hydrogen carrier and the catalyst for hydrogen production from FA has received significant attention. However, the development of efficient non-noble metal catalysts still remains challenges. In this work, we provide a technologically rather simple and environmental-friendly strategy to synthesize Co2P catalyst for dehydrogenation of FA by pyrolyzing soybean powder and cobalt salt. The K-containing solid bases in catalyst could act as Lewis acid sites for the HCOO− intermediate adsorption while the self-doped N could act as Lewis base sites to enhance the H+ adsorption. The P contained in soybean could combine with Co to form Co2P for H−C bond cleavage of HCOO−. At a Co(NO3)2·6H2O/soybean mass ratio of 1∶15, the as prepared Co2P catalyst demonstrated a gas production rate of 237.47 mL/(g·h) and a good stability. This study provides a novel strategy to develop non-noble metal heterogeneous catalysts for FA dehydrogenation.
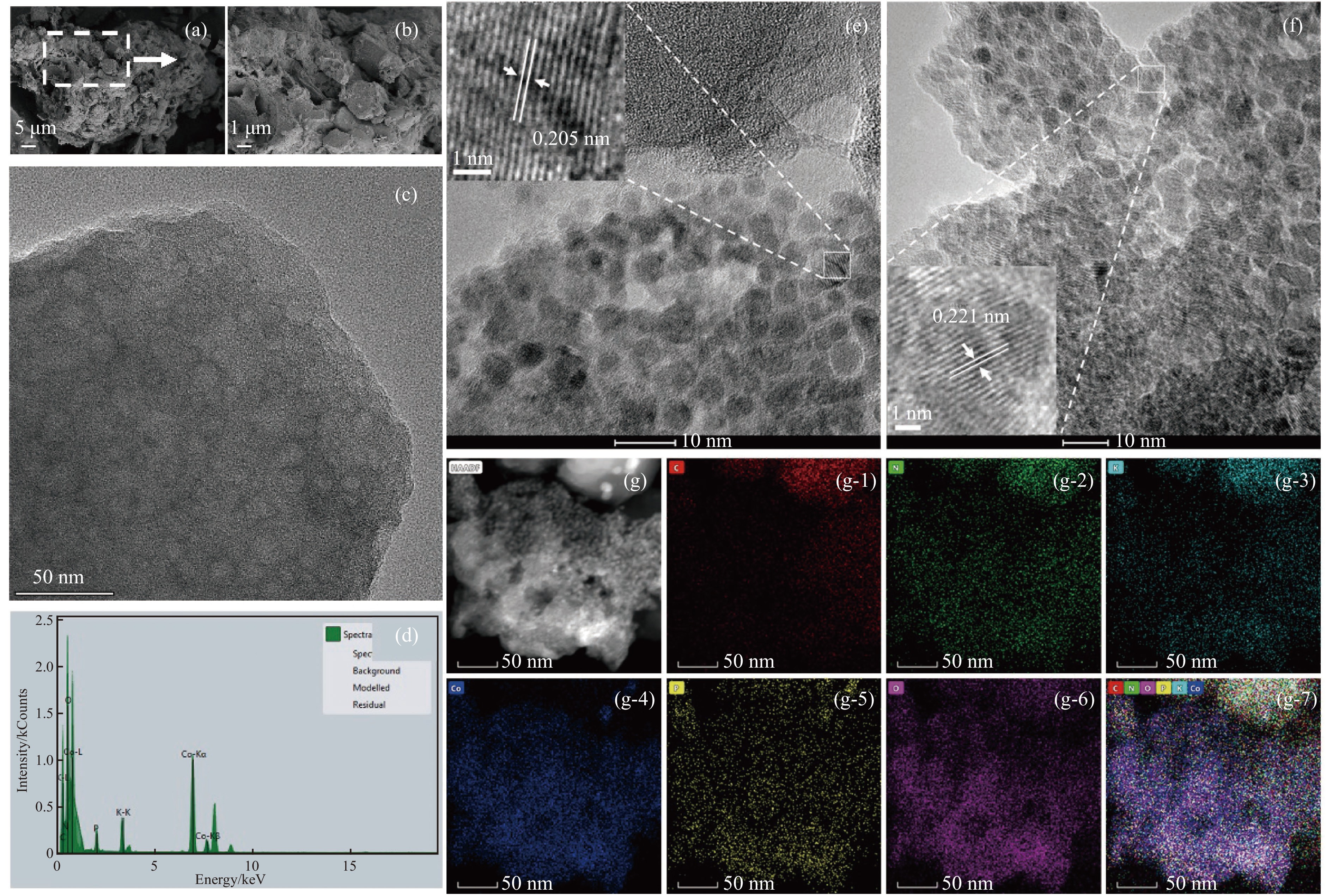
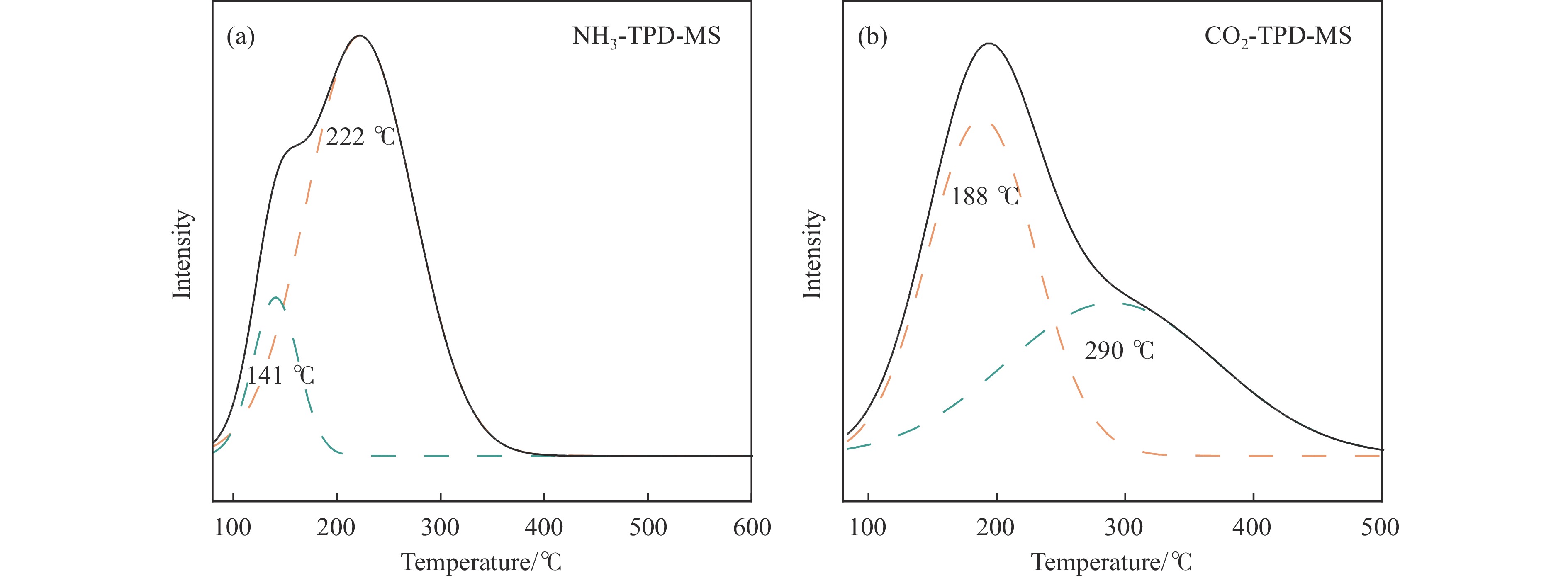
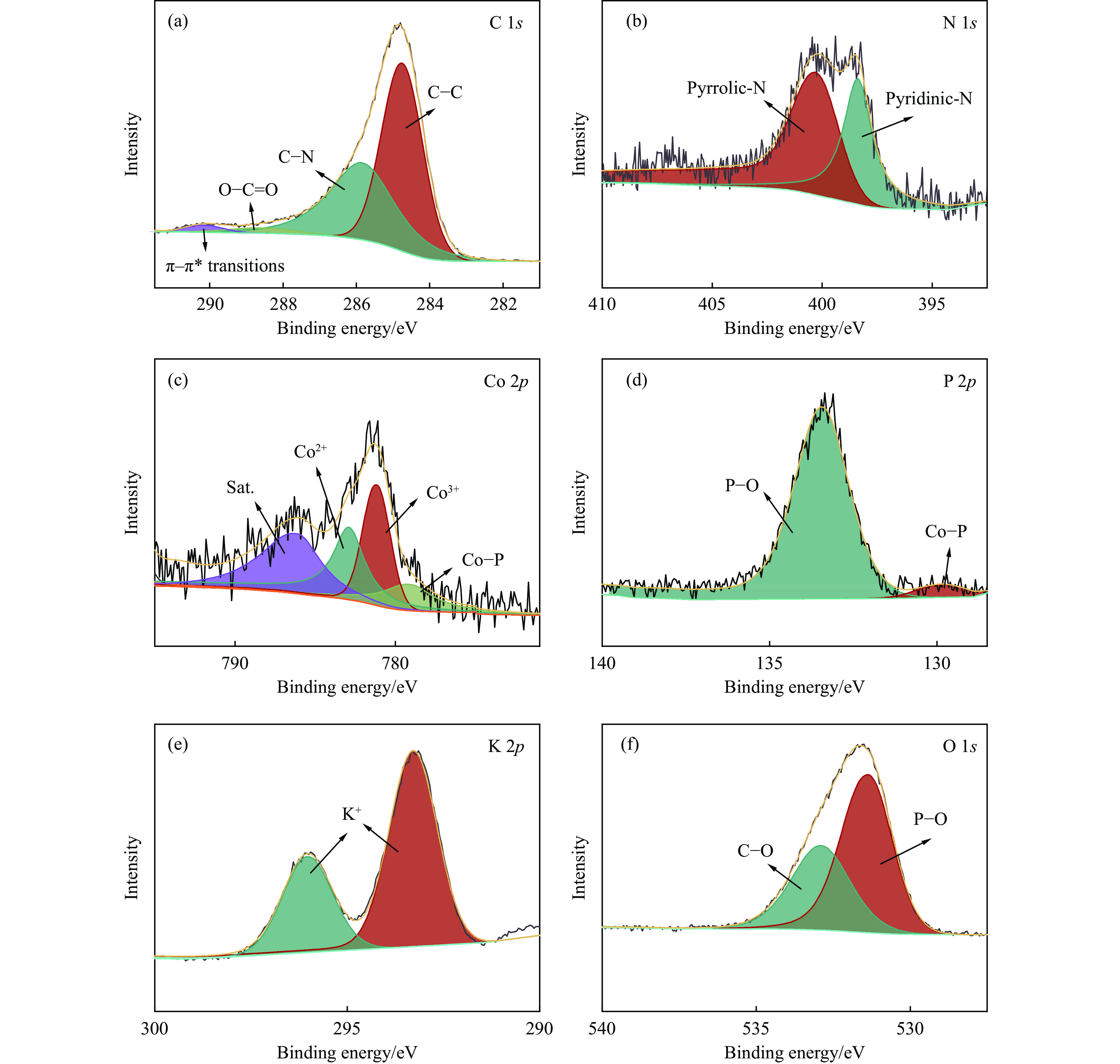

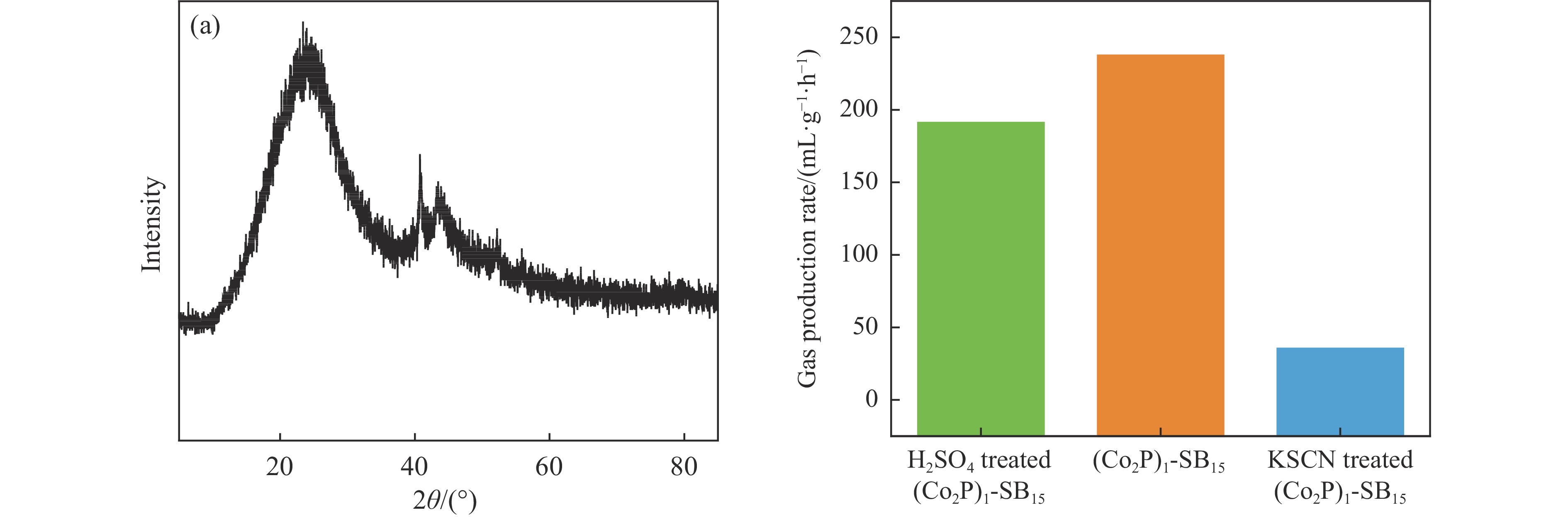
当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60433-0
摘要:
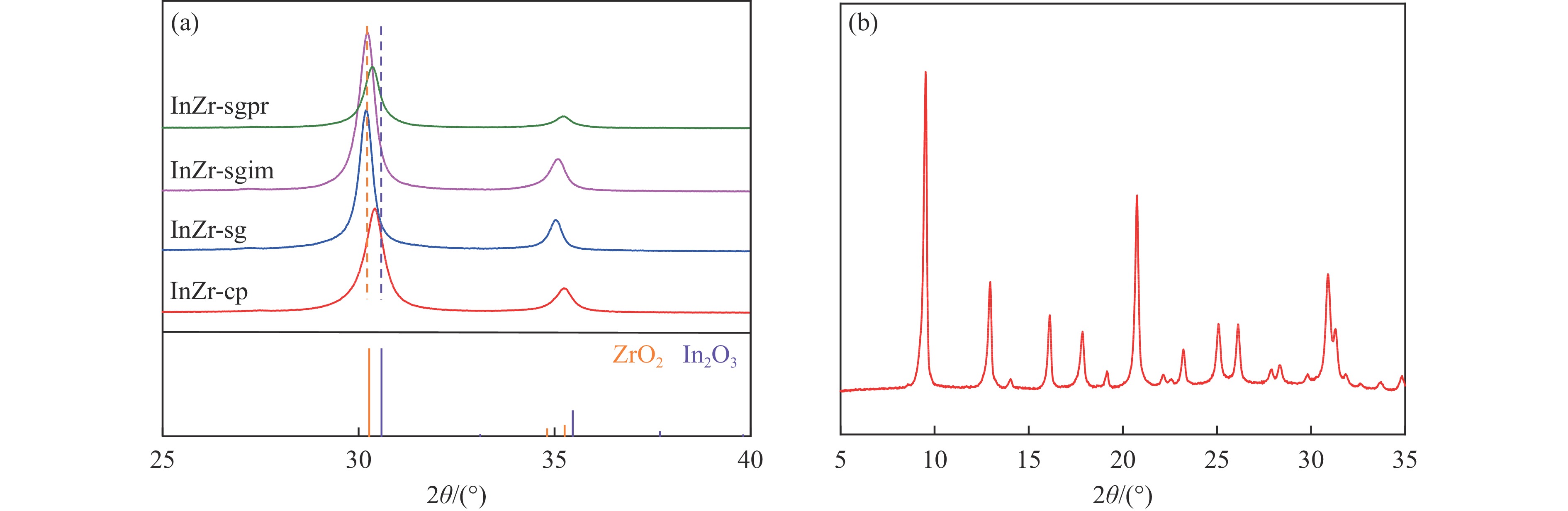
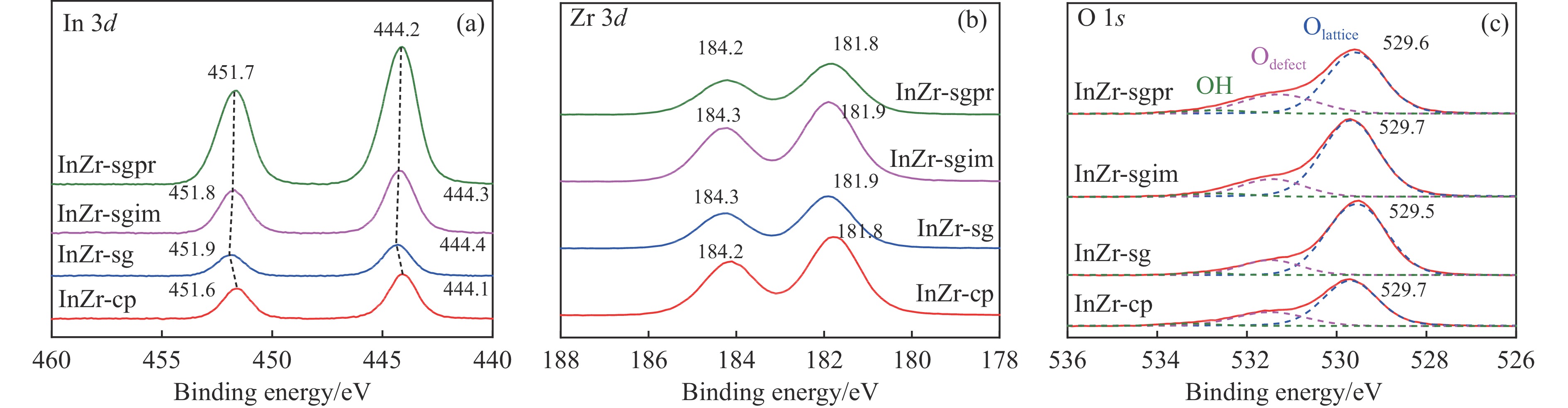
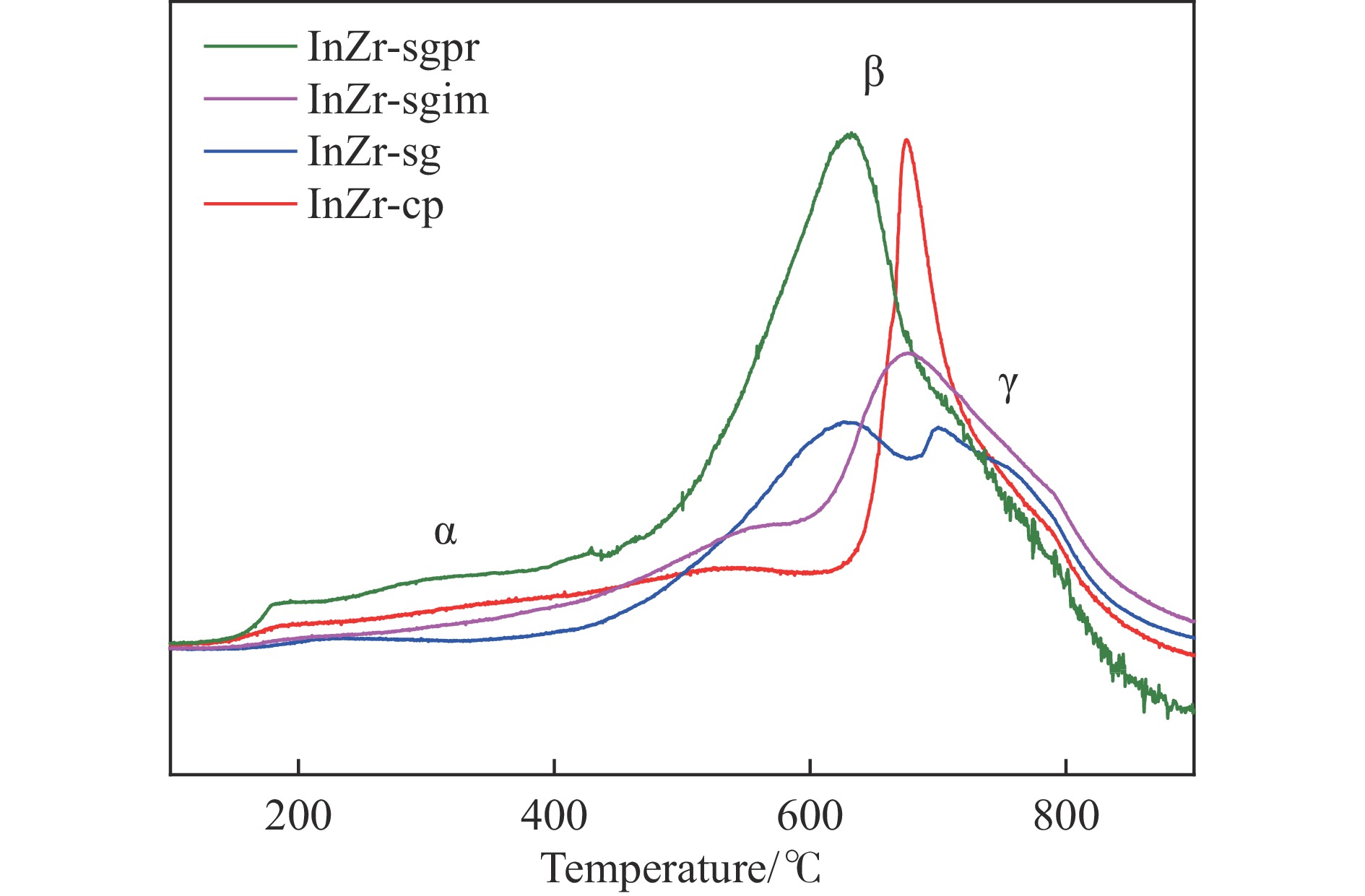
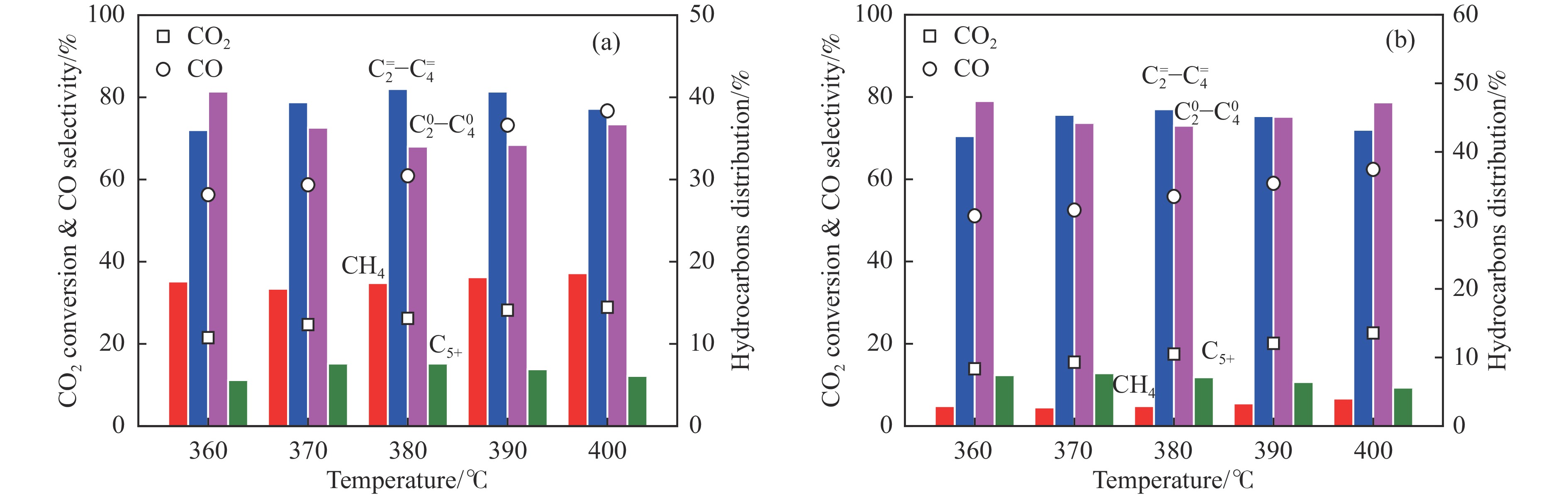
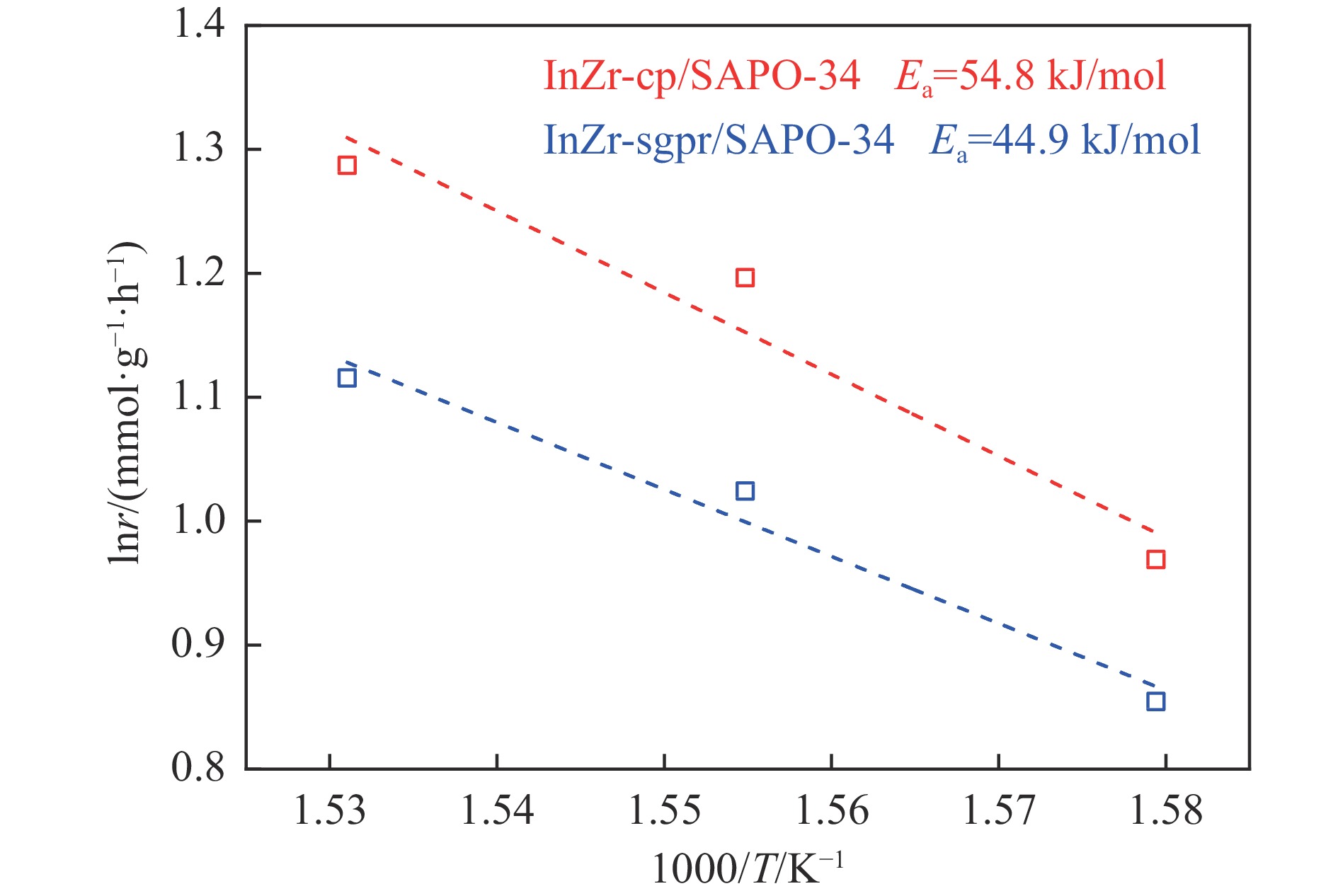
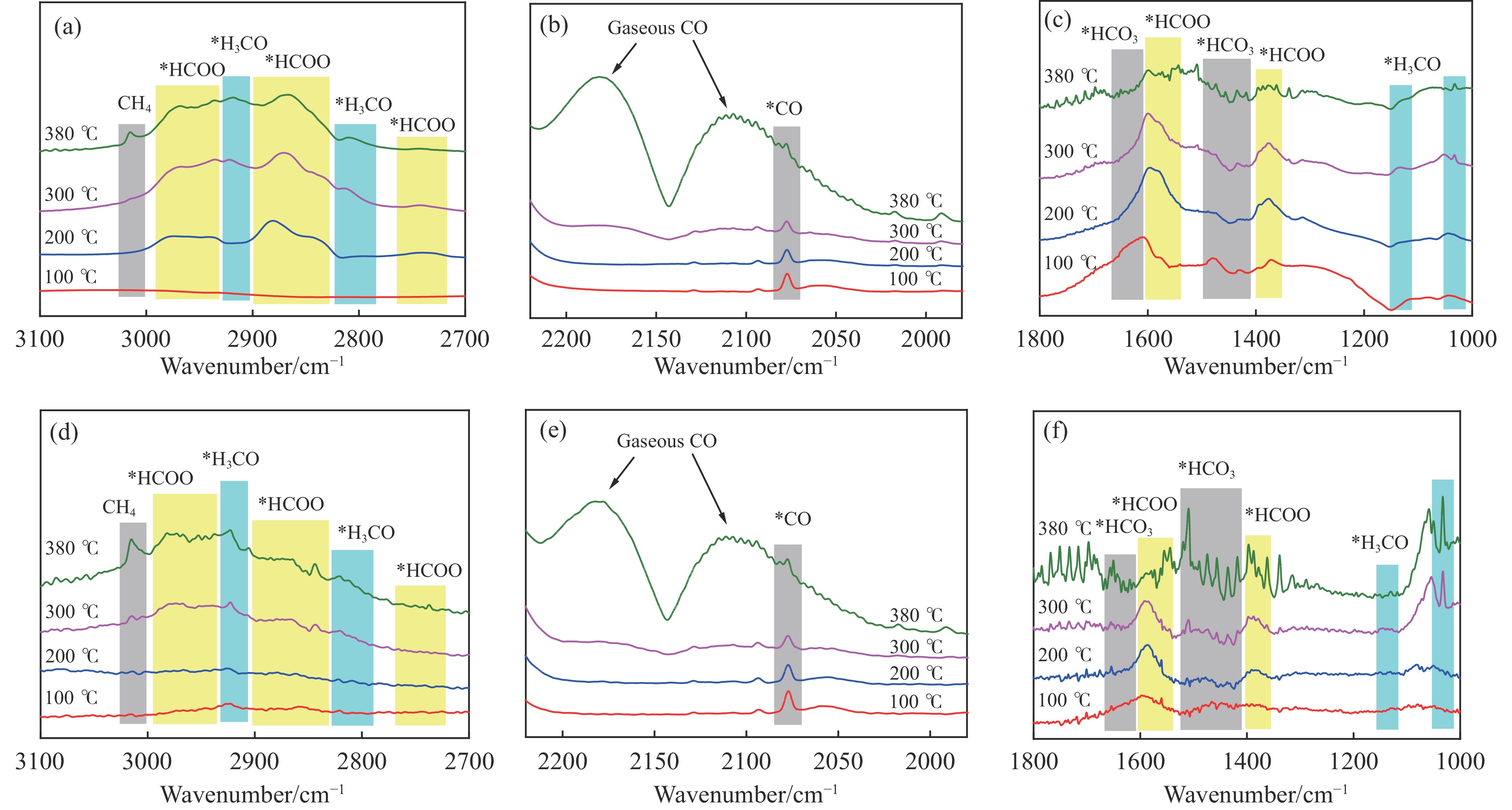
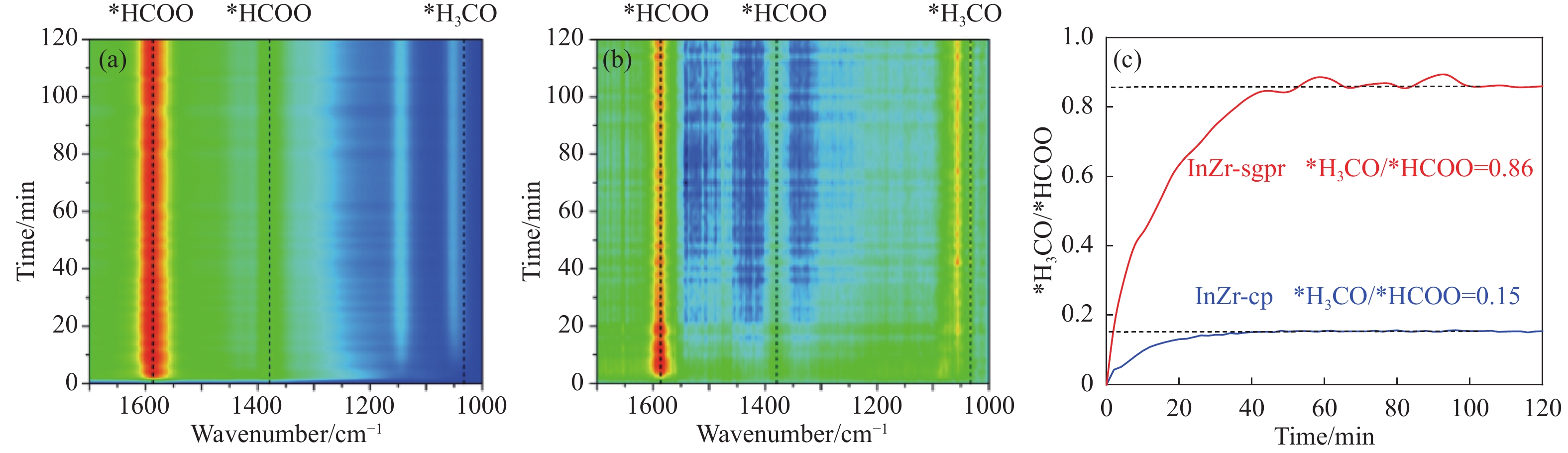
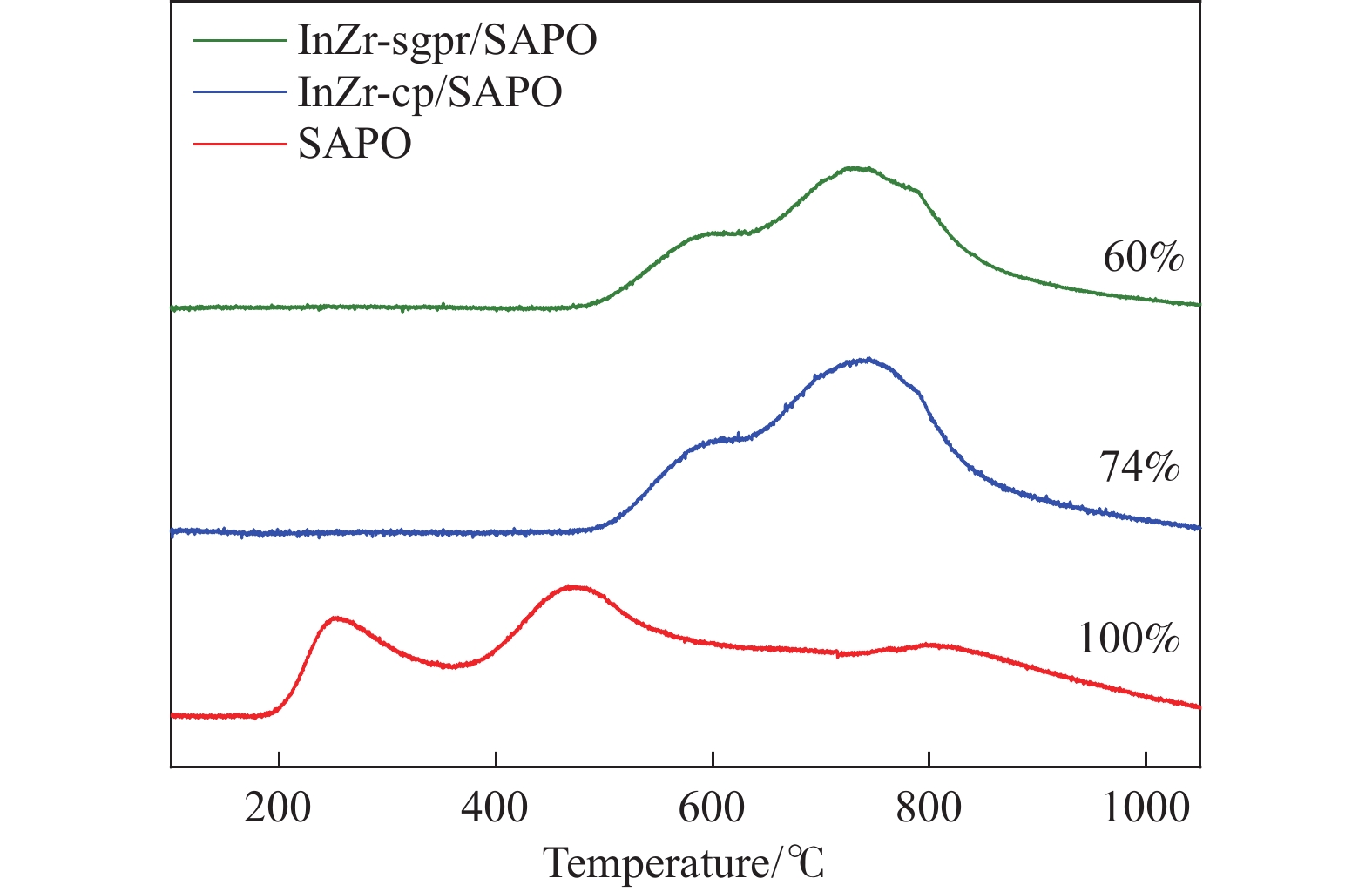
低碳烯烃是重要的化工原料,乙烯更是作为衡量一个国家石油化工发展水平的重要标志,使用CO2催化加氢制备低碳烯烃是实现CO2高值化利用的重要途径。在众多催化剂中,InZr复合氧化物/SAPO-34催化剂因其高低碳烯烃选择性和高稳定性展现出潜在的研究和应用前景。本工作研究了不同制备方法对InZr复合氧化物/SAPO-34催化剂CO2加氢制备低碳烯烃的影响,结果表明,共沉淀法制备的催化剂具有最高的催化活性,溶胶凝胶-沉积法制备的催化剂具有最高的低碳烯烃选择性,并结合多种表征手段揭示了InZr复合氧化物/SAPO-34催化剂内在构效关系。
低碳烯烃是重要的化工原料,乙烯更是作为衡量一个国家石油化工发展水平的重要标志,使用CO2催化加氢制备低碳烯烃是实现CO2高值化利用的重要途径。在众多催化剂中,InZr复合氧化物/SAPO-34催化剂因其高低碳烯烃选择性和高稳定性展现出潜在的研究和应用前景。本工作研究了不同制备方法对InZr复合氧化物/SAPO-34催化剂CO2加氢制备低碳烯烃的影响,结果表明,共沉淀法制备的催化剂具有最高的催化活性,溶胶凝胶-沉积法制备的催化剂具有最高的低碳烯烃选择性,并结合多种表征手段揭示了InZr复合氧化物/SAPO-34催化剂内在构效关系。

当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60412-8
摘要:
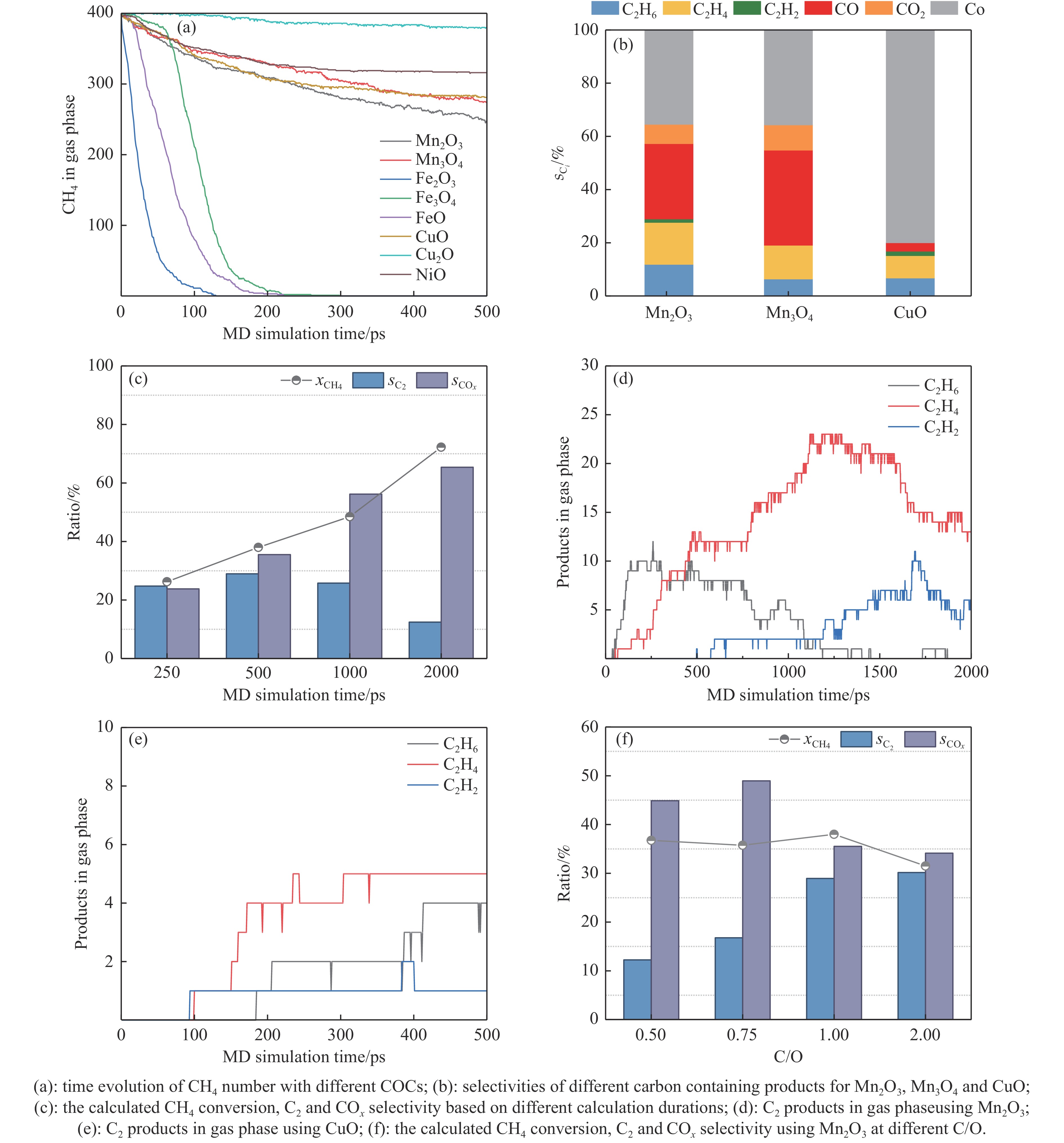
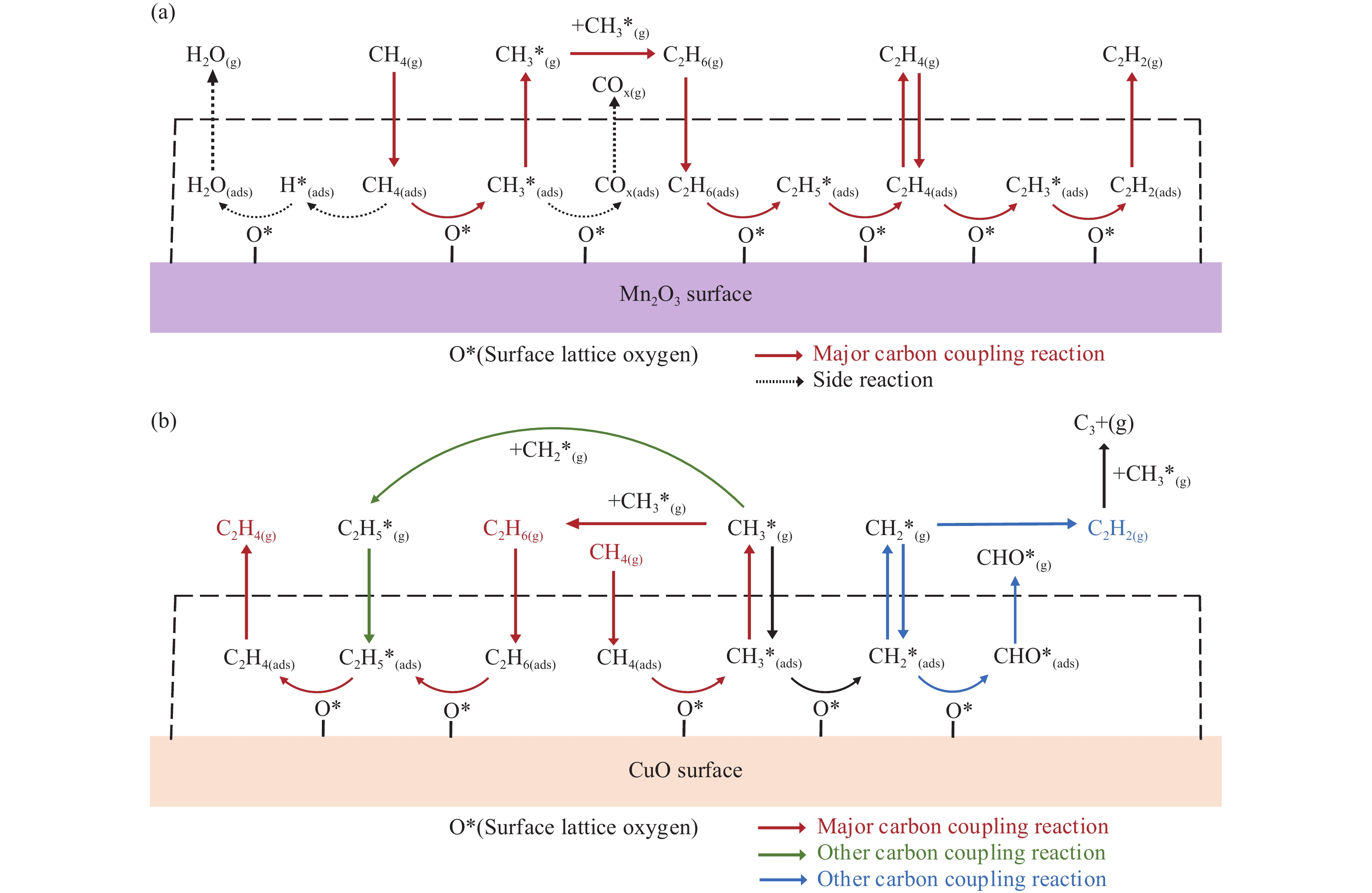
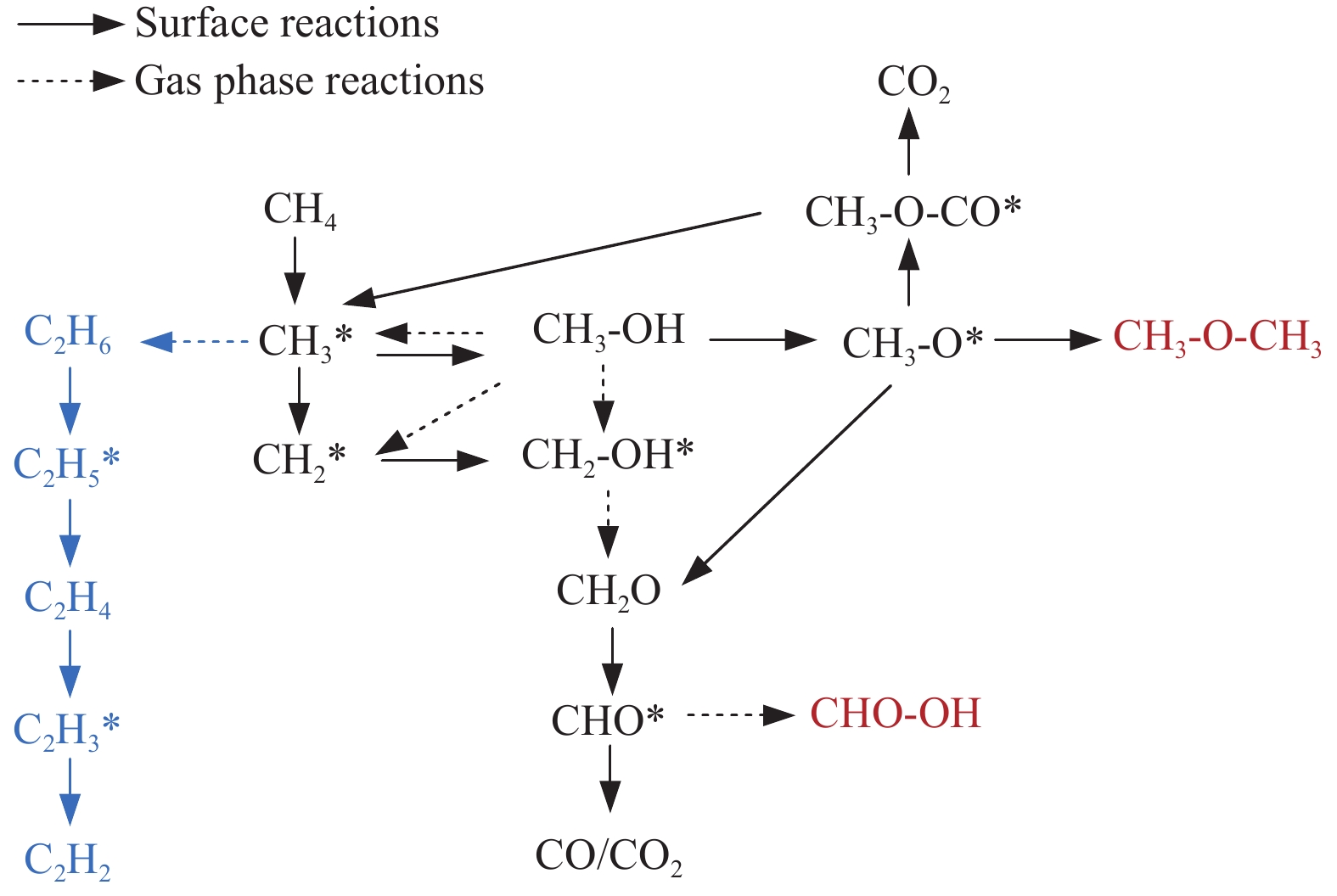
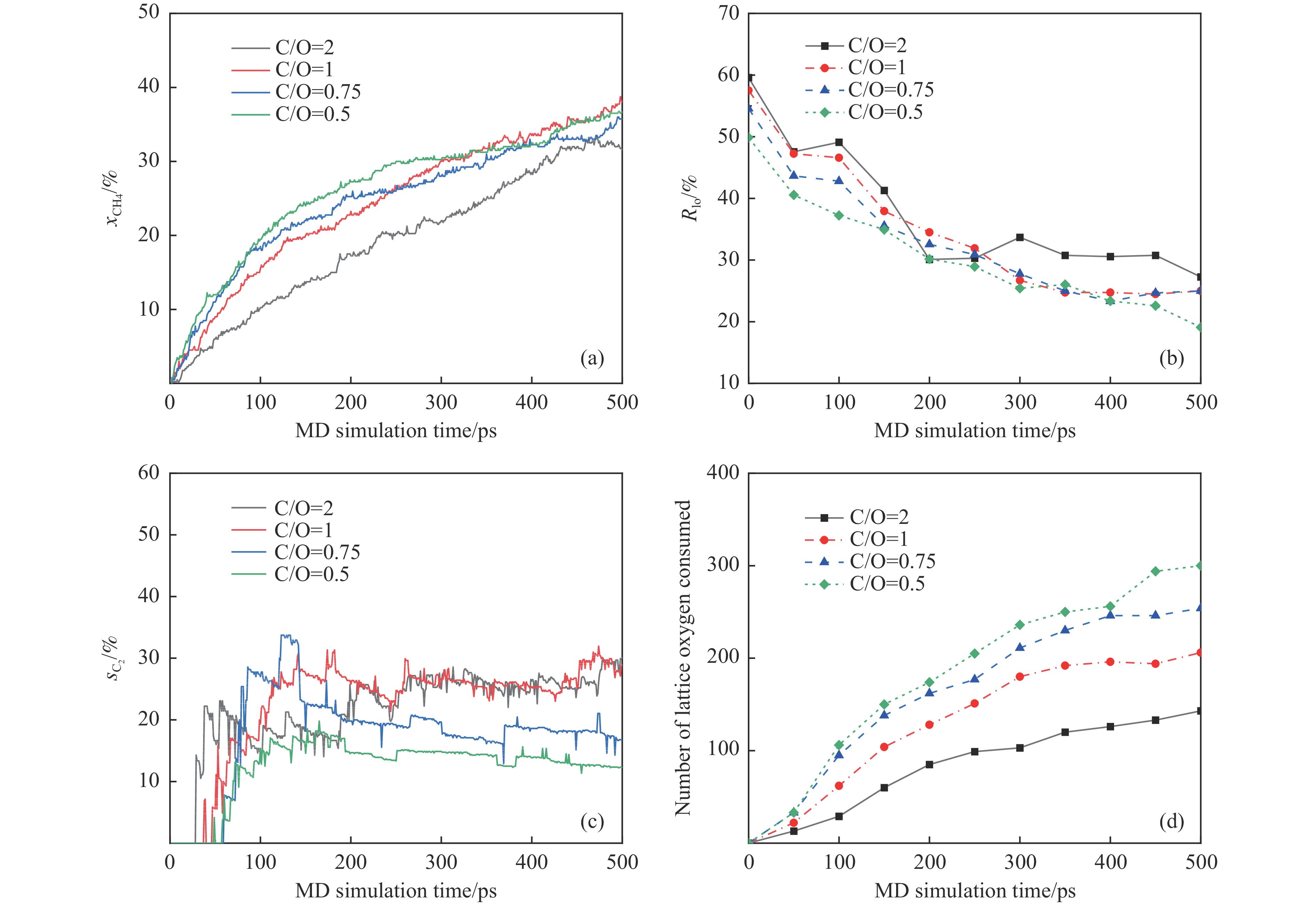
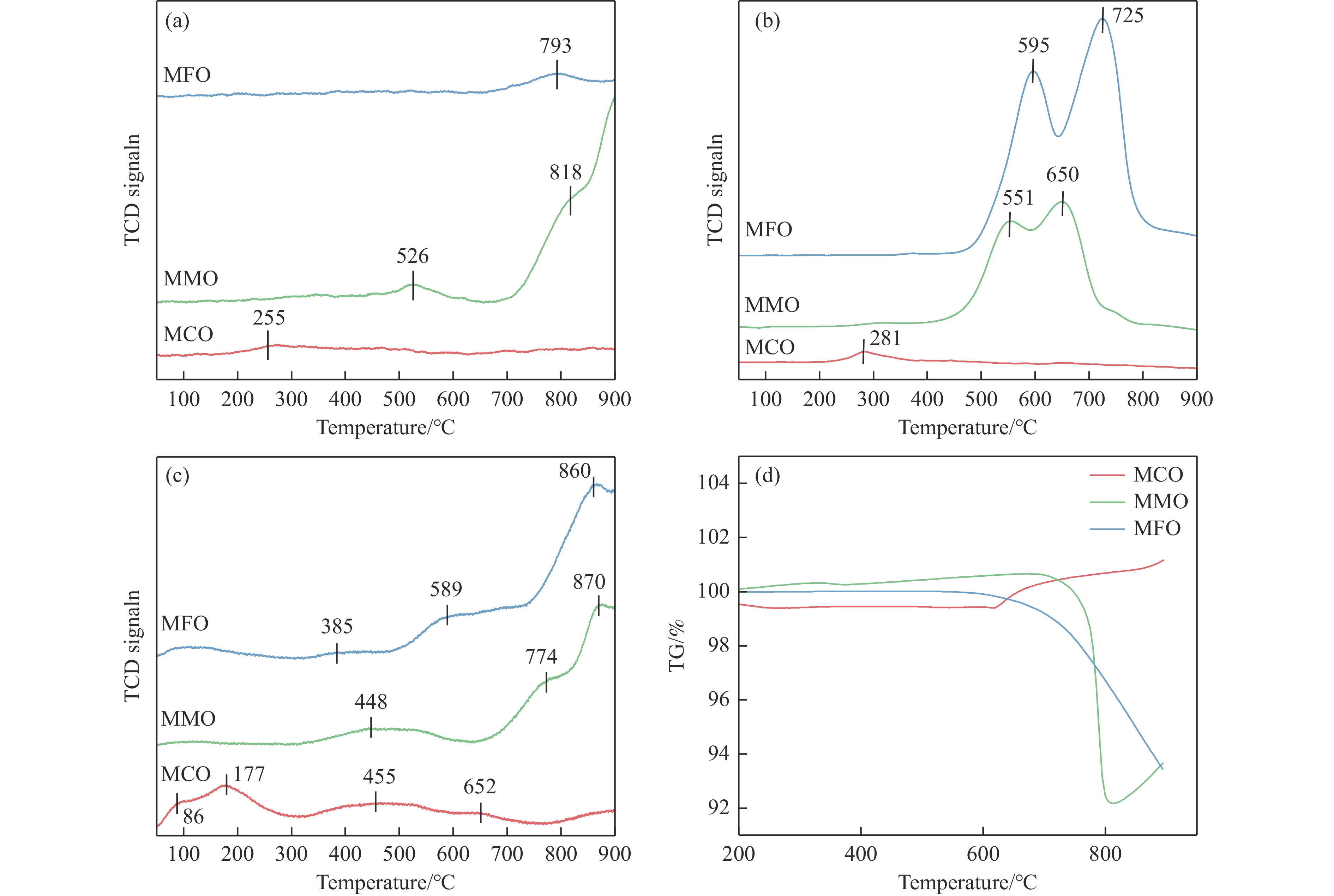
本研究采用分子动力学模拟的方法计算八种金属氧化物催化剂-载氧体CL-OCM反应性能,并对性能最优的Mn2O3开展反应时间和颗粒尺寸的研究。结果表明,适当延长反应时间有利于提高 C2H4 选择性; C/O=1 是Mn2O3的理想尺寸。基于以上结果分析了Mn2O3 CL-OCM界面反应路径和晶格氧传递问题,以揭示反应机理。CH3 *气相二聚化生成C2H6的是CL-OCM最主要的碳偶联路径。除此之外,还存在两条碳偶联路径,均由CH2 *引发。CH3 *与OH*表面结合生成甲醇是CL-OCM副反应的先决步骤,抑制甲醇生成是提高CL-OCM反应C2选择性的关键。晶格氧存在转化,表面晶格氧是甲烷活化的活性氧。晶格氧数量差异及体相晶格氧迁移阻力差异是导致CH4转化率和C2选择性不同的主要原因。该研究为CL-OCM催化剂-载氧体的机理探究提供新的方法。
本研究采用分子动力学模拟的方法计算八种金属氧化物催化剂-载氧体CL-OCM反应性能,并对性能最优的Mn2O3开展反应时间和颗粒尺寸的研究。结果表明,适当延长反应时间有利于提高 C2H4 选择性; C/O=1 是Mn2O3的理想尺寸。基于以上结果分析了Mn2O3 CL-OCM界面反应路径和晶格氧传递问题,以揭示反应机理。CH3 *气相二聚化生成C2H6的是CL-OCM最主要的碳偶联路径。除此之外,还存在两条碳偶联路径,均由CH2 *引发。CH3 *与OH*表面结合生成甲醇是CL-OCM副反应的先决步骤,抑制甲醇生成是提高CL-OCM反应C2选择性的关键。晶格氧存在转化,表面晶格氧是甲烷活化的活性氧。晶格氧数量差异及体相晶格氧迁移阻力差异是导致CH4转化率和C2选择性不同的主要原因。该研究为CL-OCM催化剂-载氧体的机理探究提供新的方法。
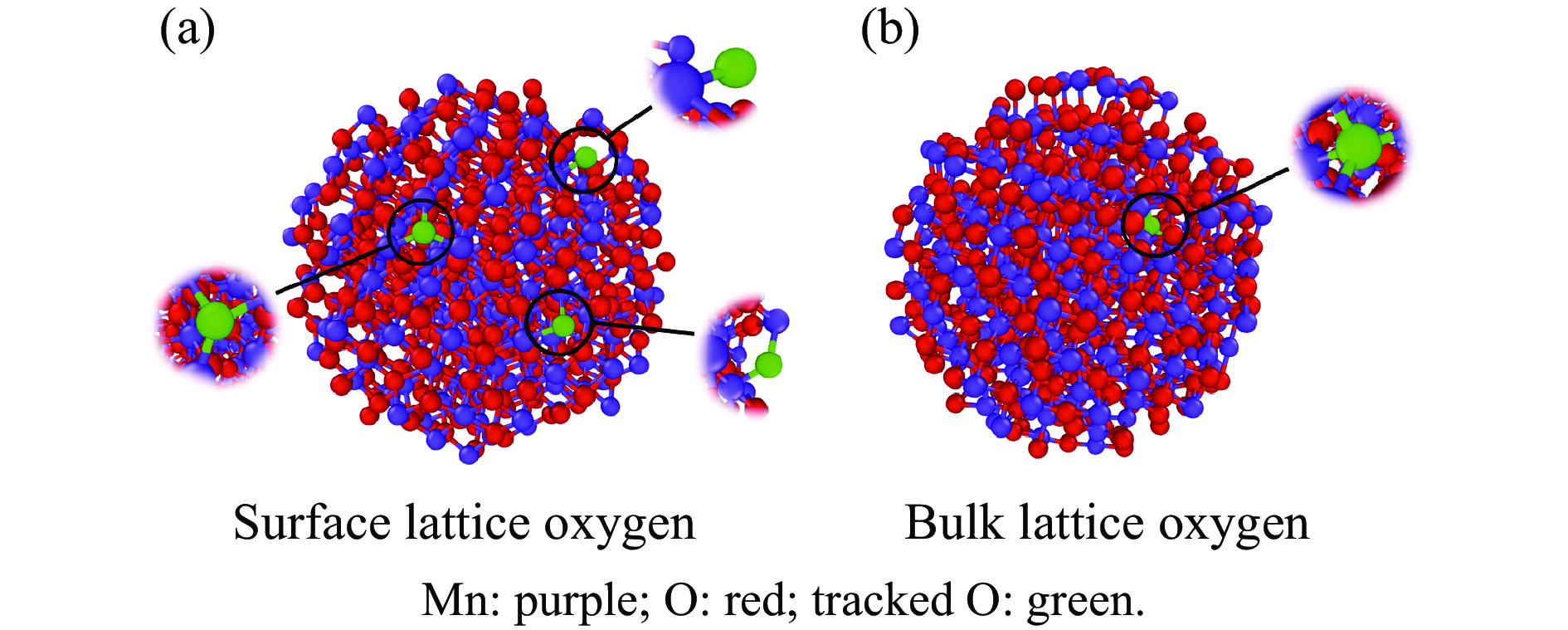

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023082
摘要:
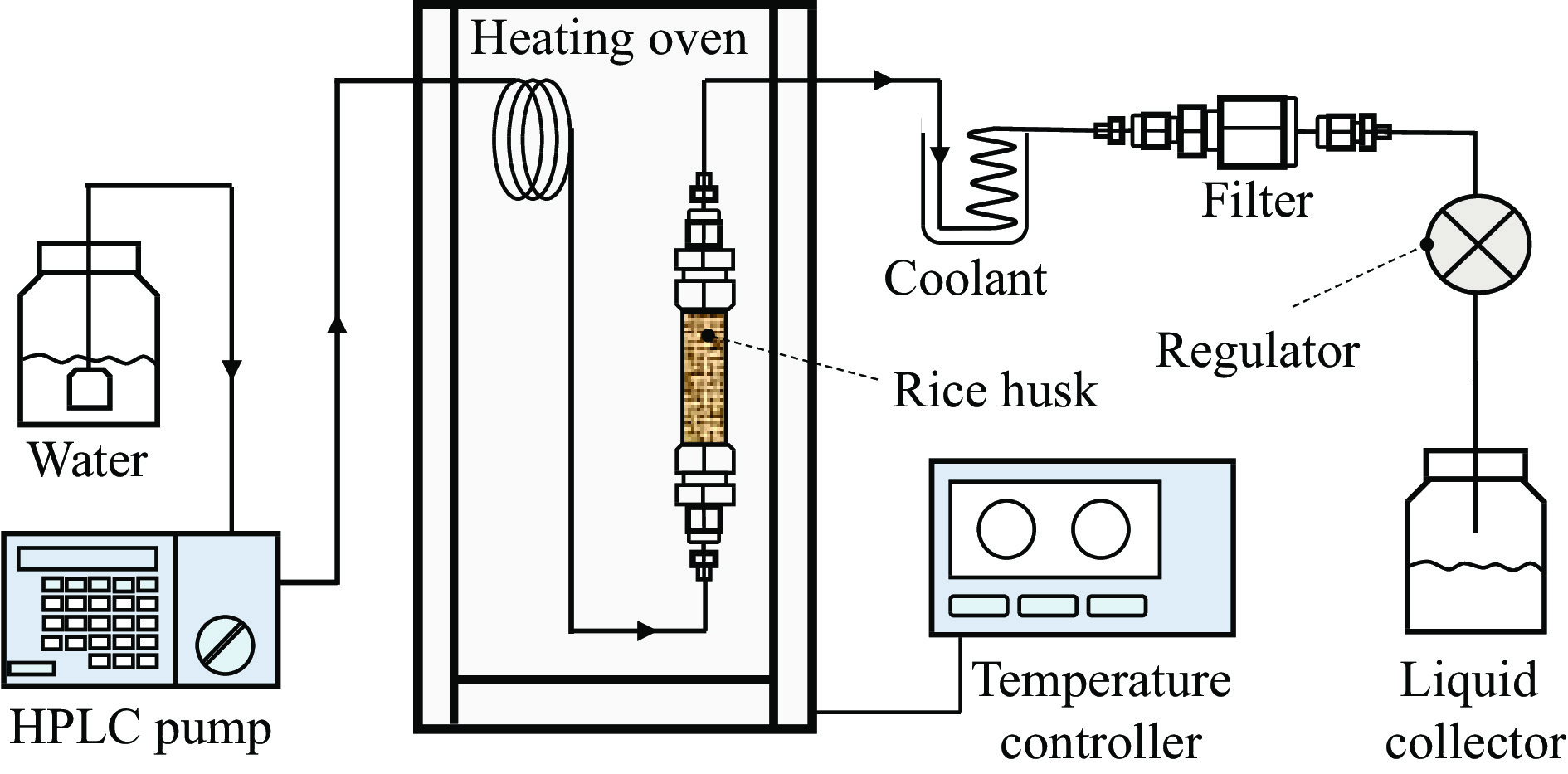
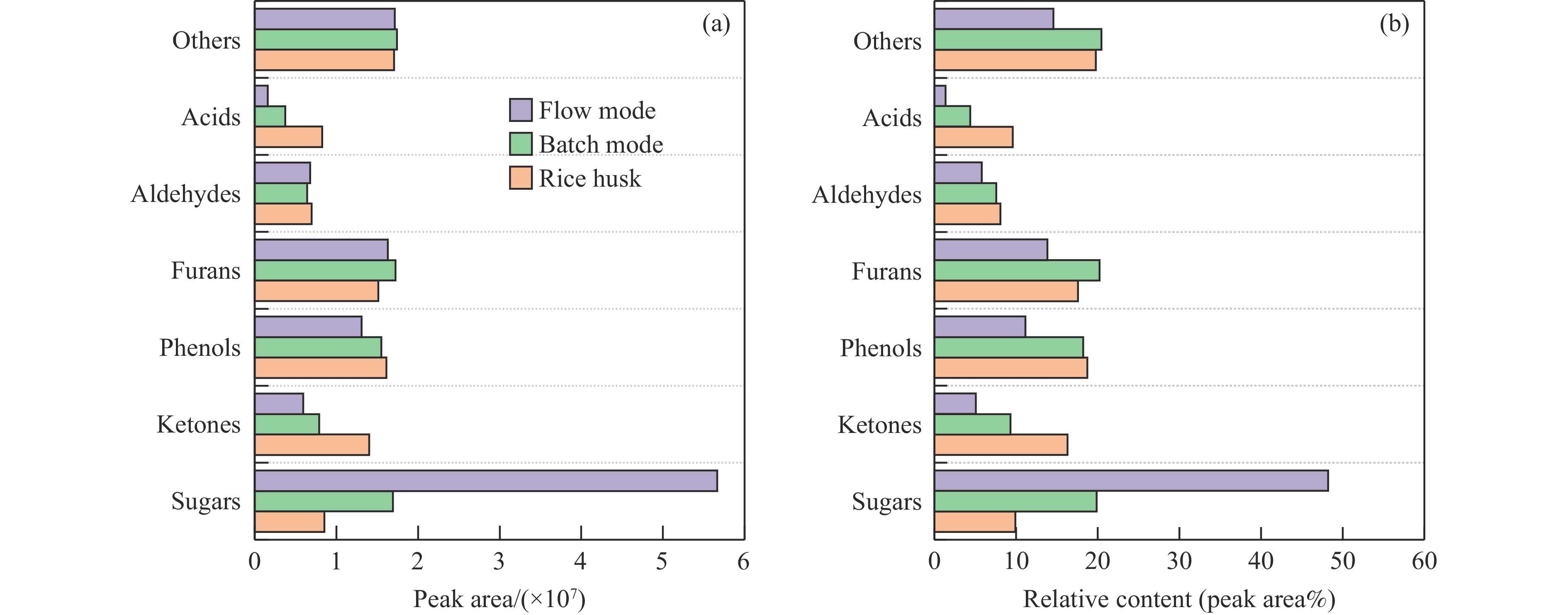
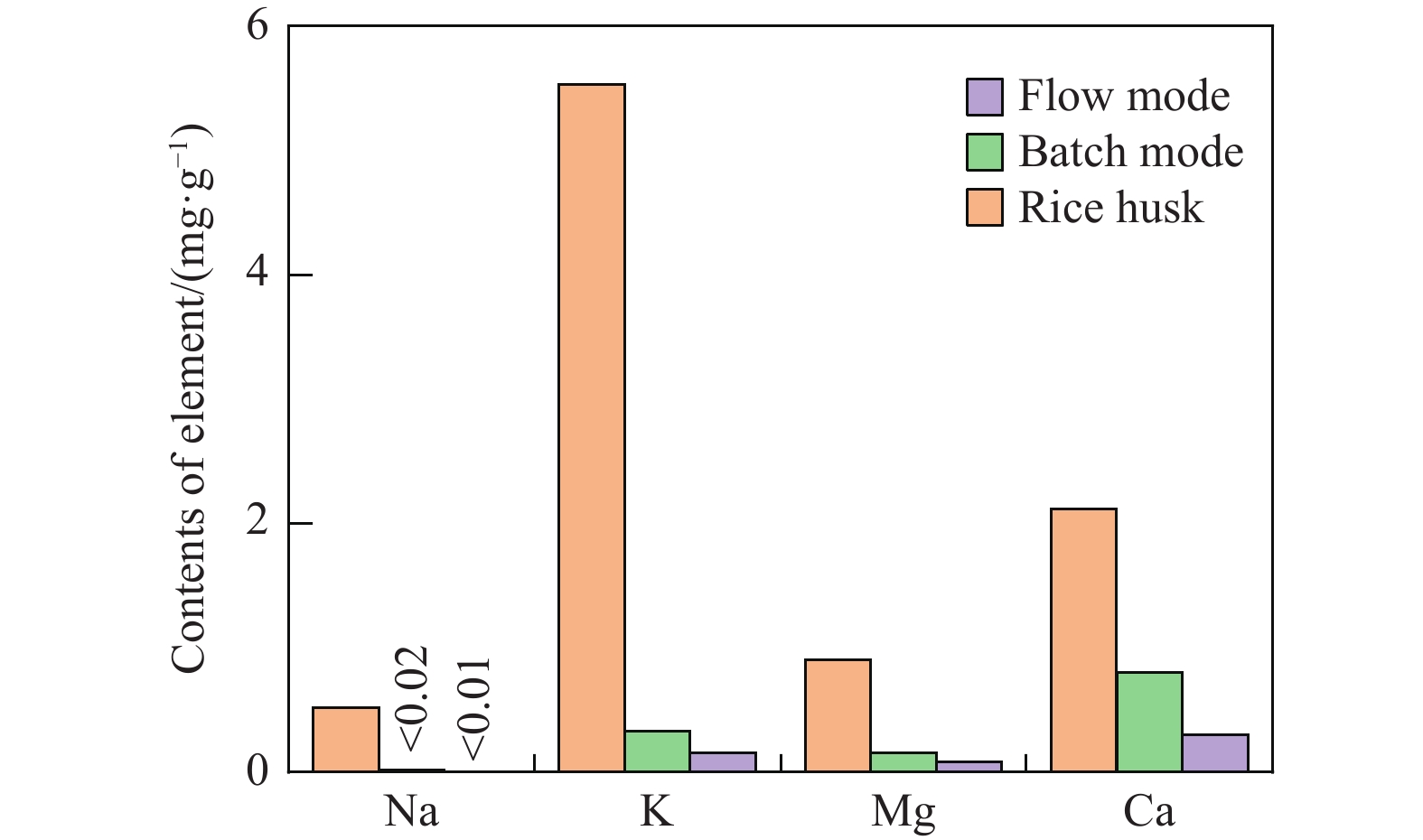
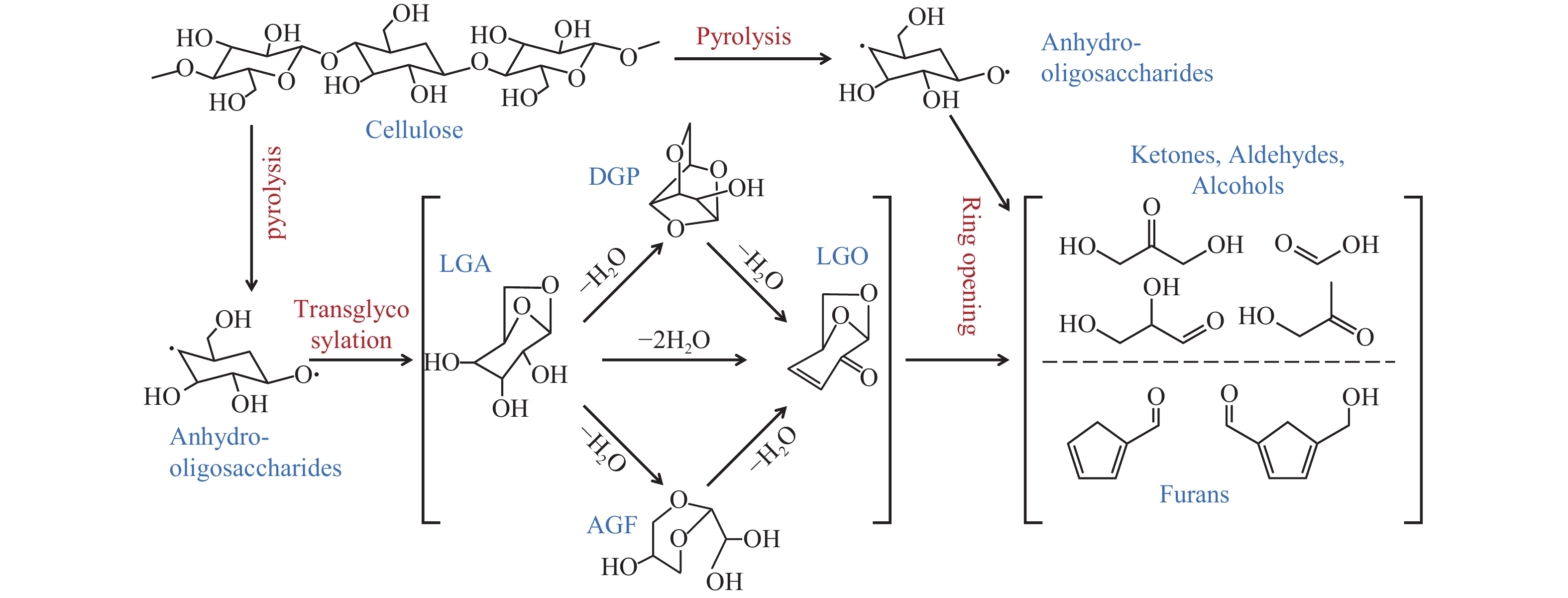
生物质复杂的多组分体系和致密交联的化学结构是制约其高值化利用的关键,实现木质纤维组分预分离对生物质分级转化具有重要意义。实验采用连续式水热法预处理稻壳,考察了水热温度和流量对稻壳分解速率以及固相产物化学组成与热解特性的影响。结果表明,稻壳的水热分解符合表面化学反应过程控制的未反应收缩核模型,预处理在180 ℃下能脱除稻壳95%的碱及碱土金属、92%的半纤维素和59%的木质素,极大保留了纤维素组分,这使得稻壳热解产物中以左旋葡聚糖为主的脱水糖的相对含量从9.9%提高至48.2%。
生物质复杂的多组分体系和致密交联的化学结构是制约其高值化利用的关键,实现木质纤维组分预分离对生物质分级转化具有重要意义。实验采用连续式水热法预处理稻壳,考察了水热温度和流量对稻壳分解速率以及固相产物化学组成与热解特性的影响。结果表明,稻壳的水热分解符合表面化学反应过程控制的未反应收缩核模型,预处理在180 ℃下能脱除稻壳95%的碱及碱土金属、92%的半纤维素和59%的木质素,极大保留了纤维素组分,这使得稻壳热解产物中以左旋葡聚糖为主的脱水糖的相对含量从9.9%提高至48.2%。
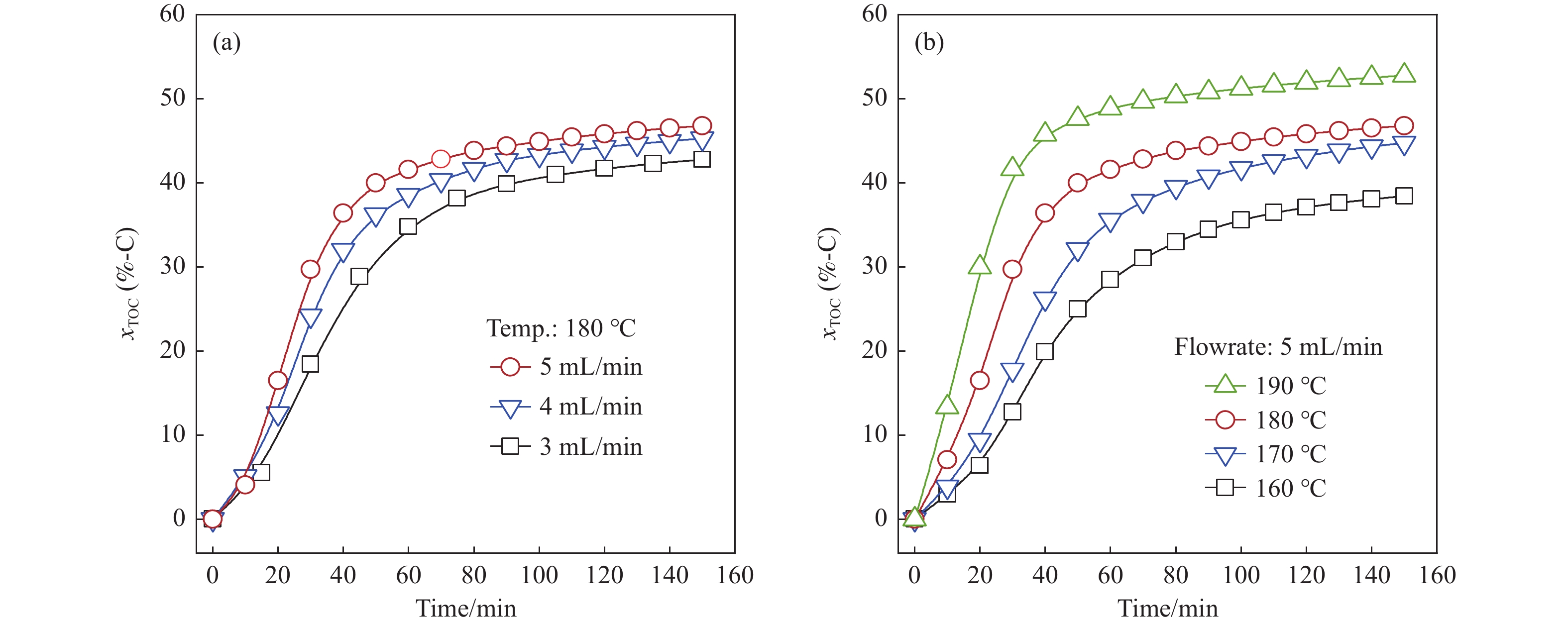
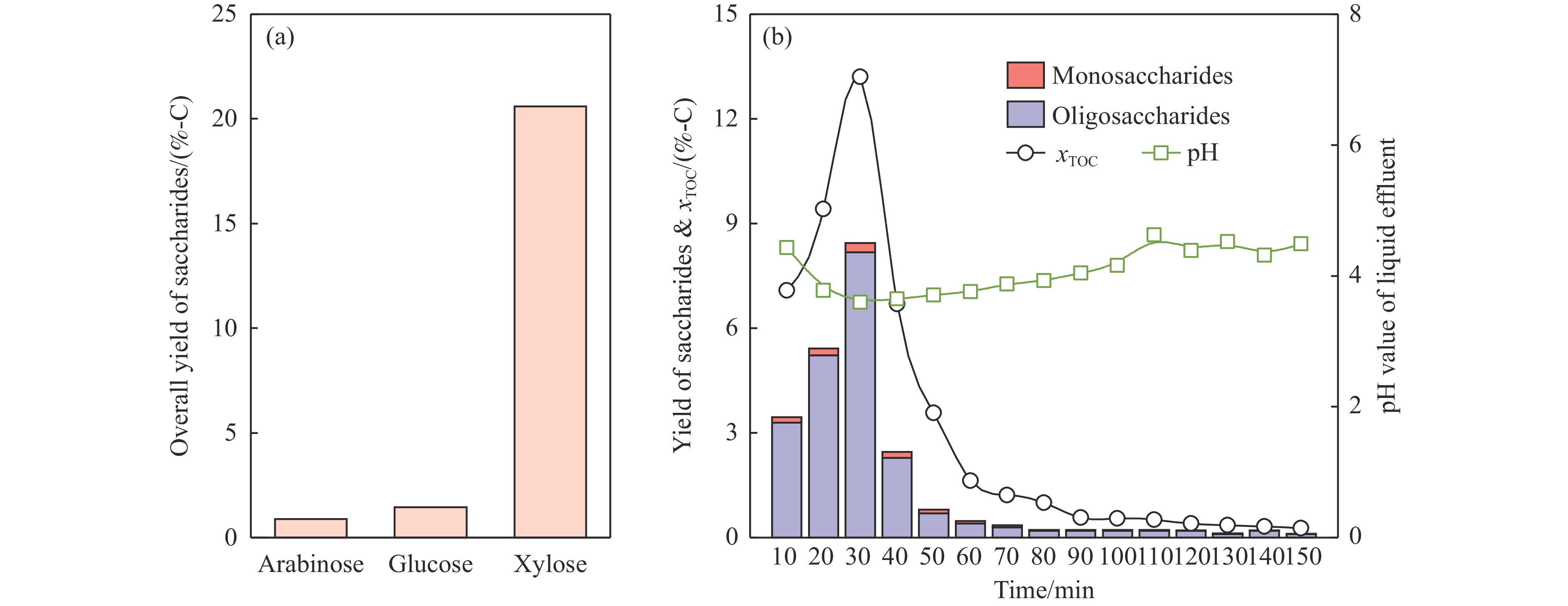

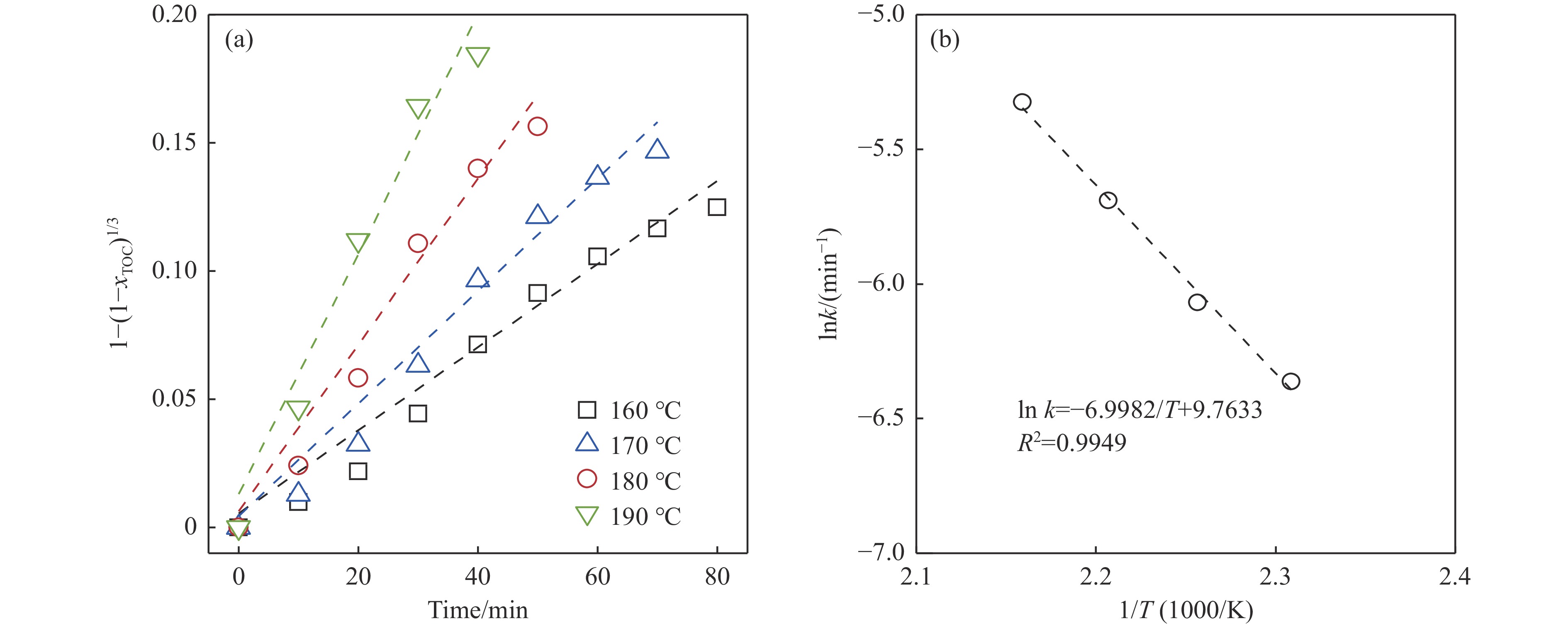
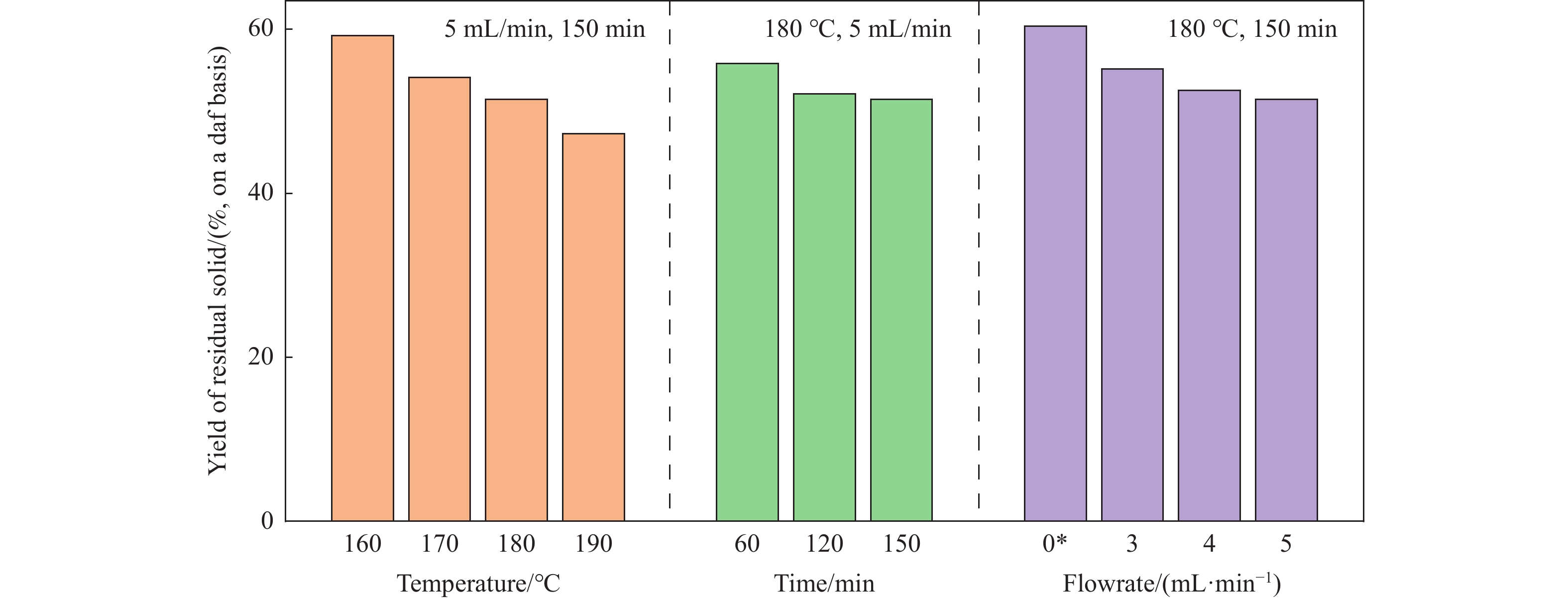

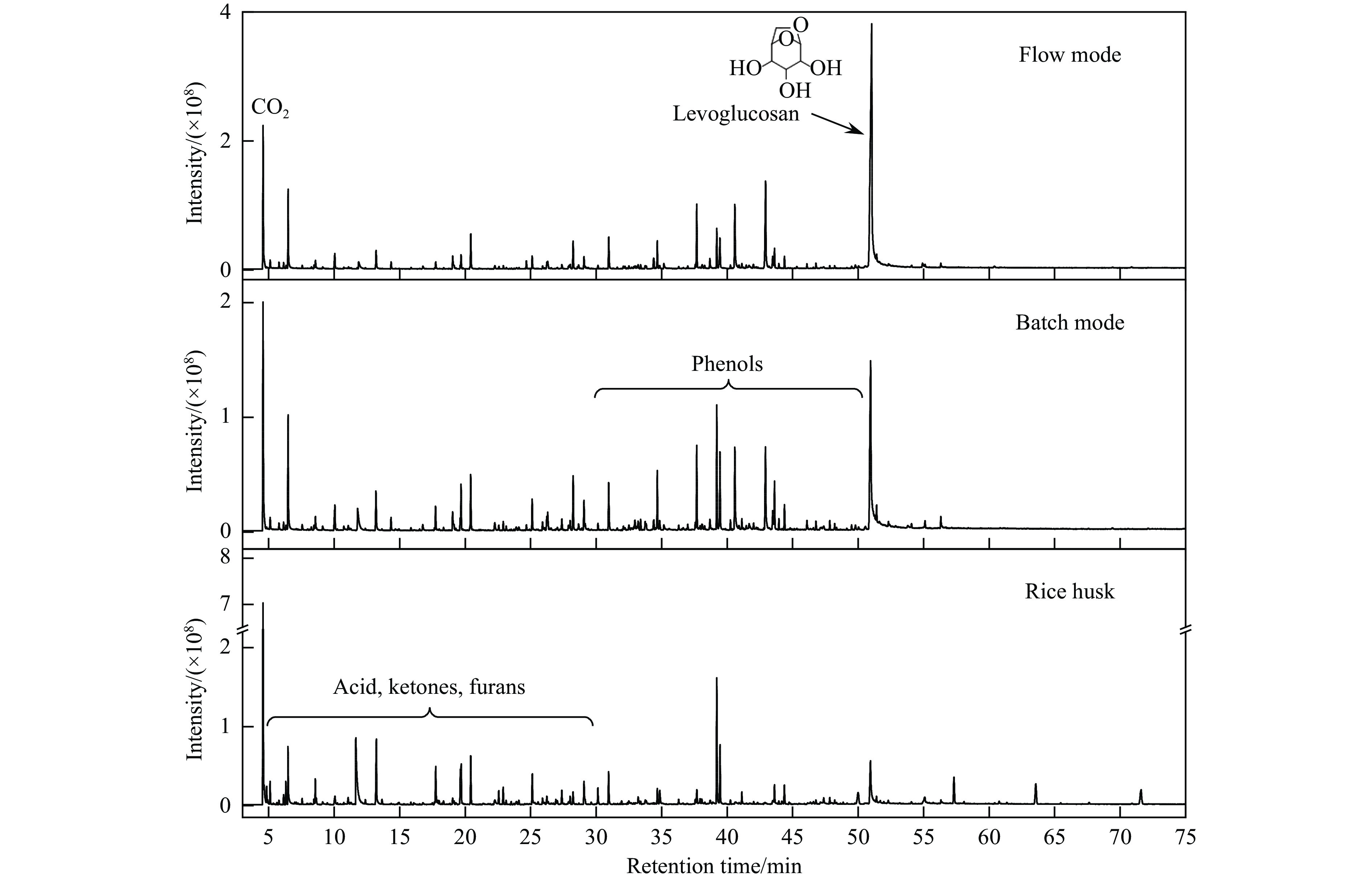
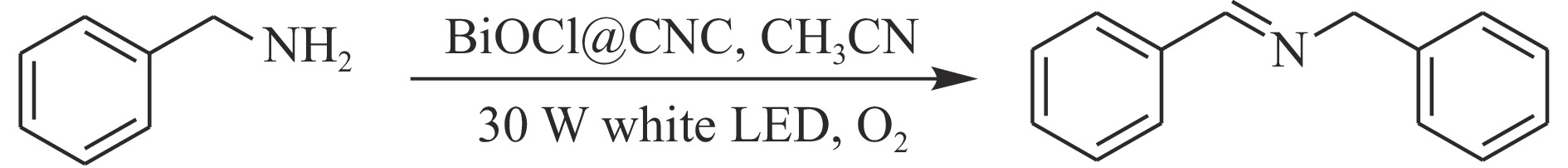
当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60437-8
摘要:
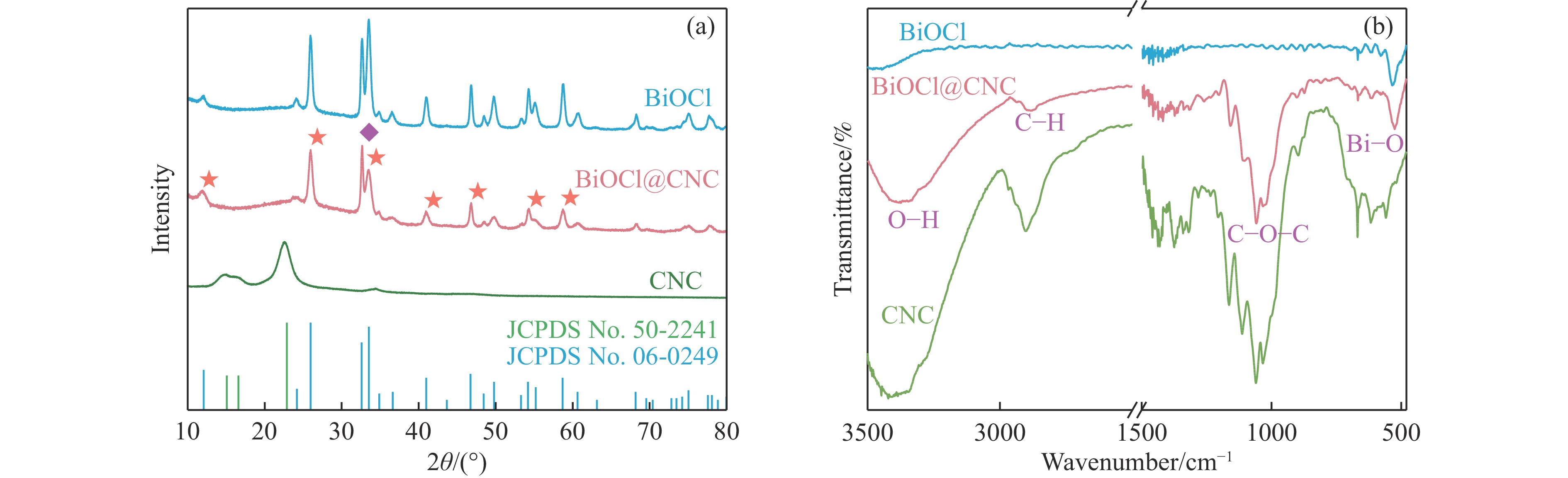
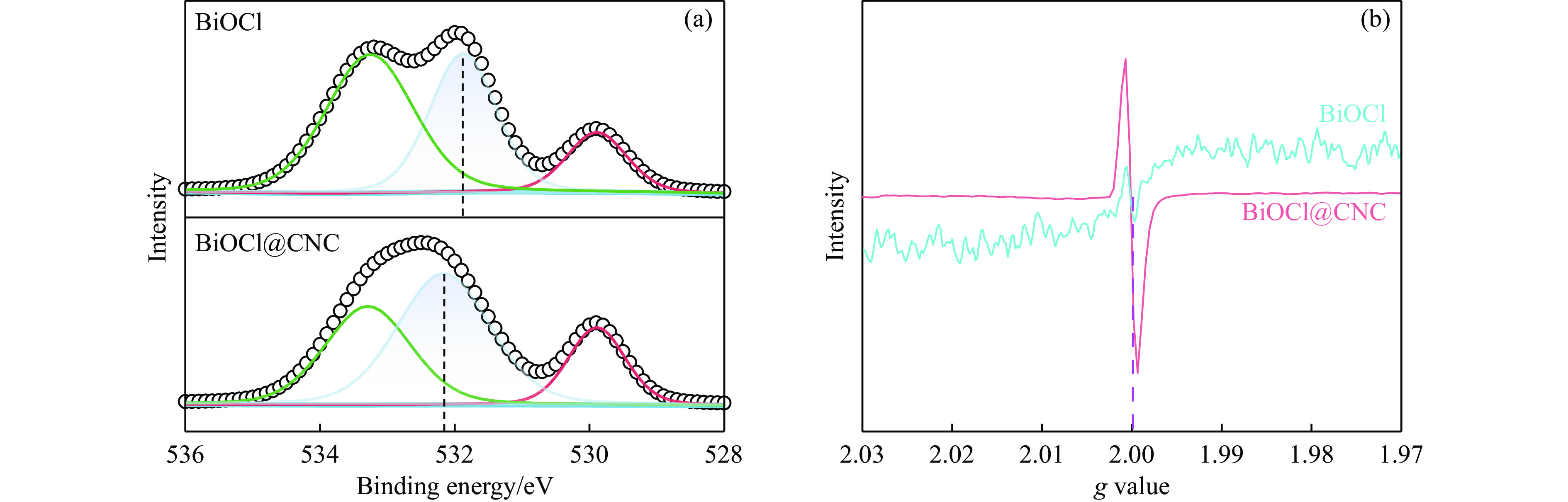
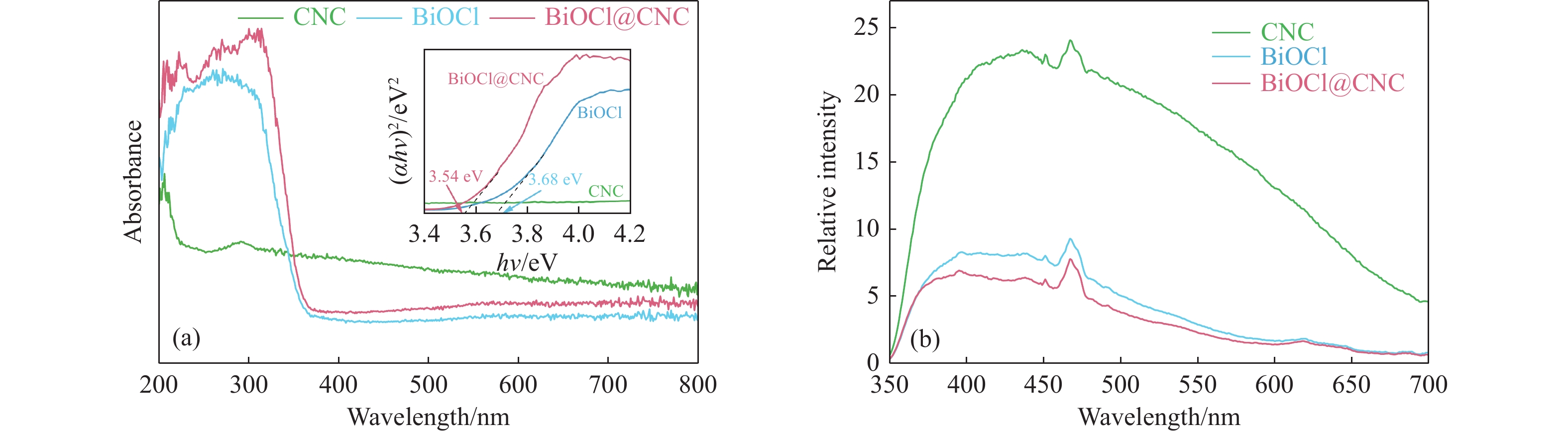
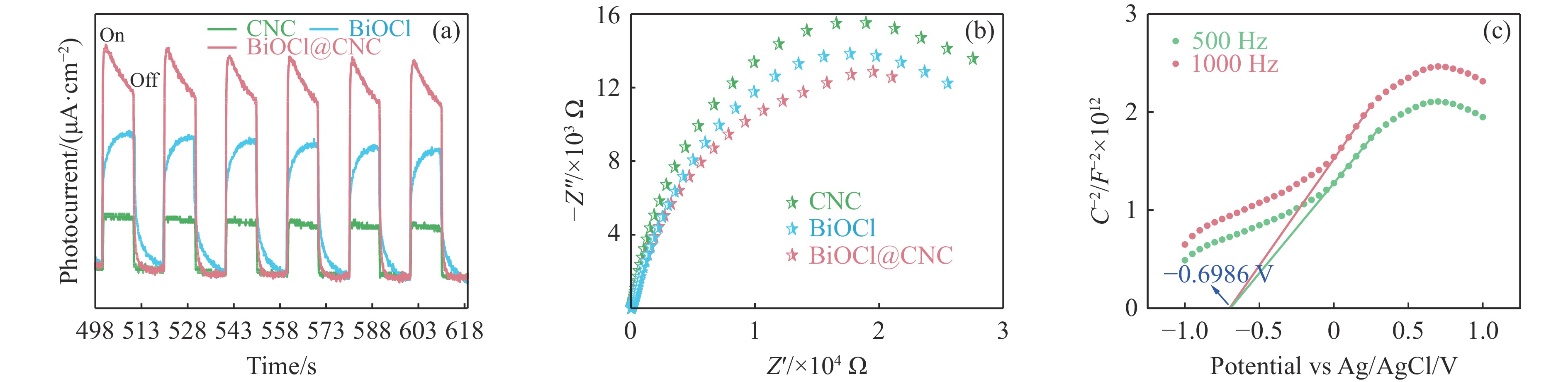
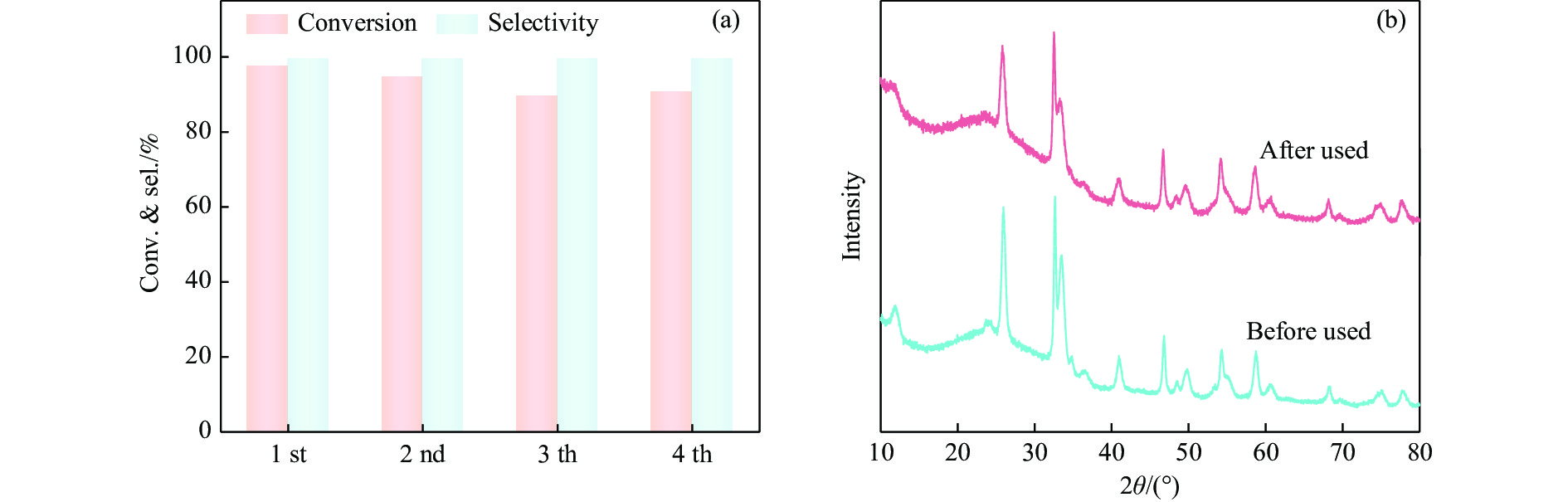
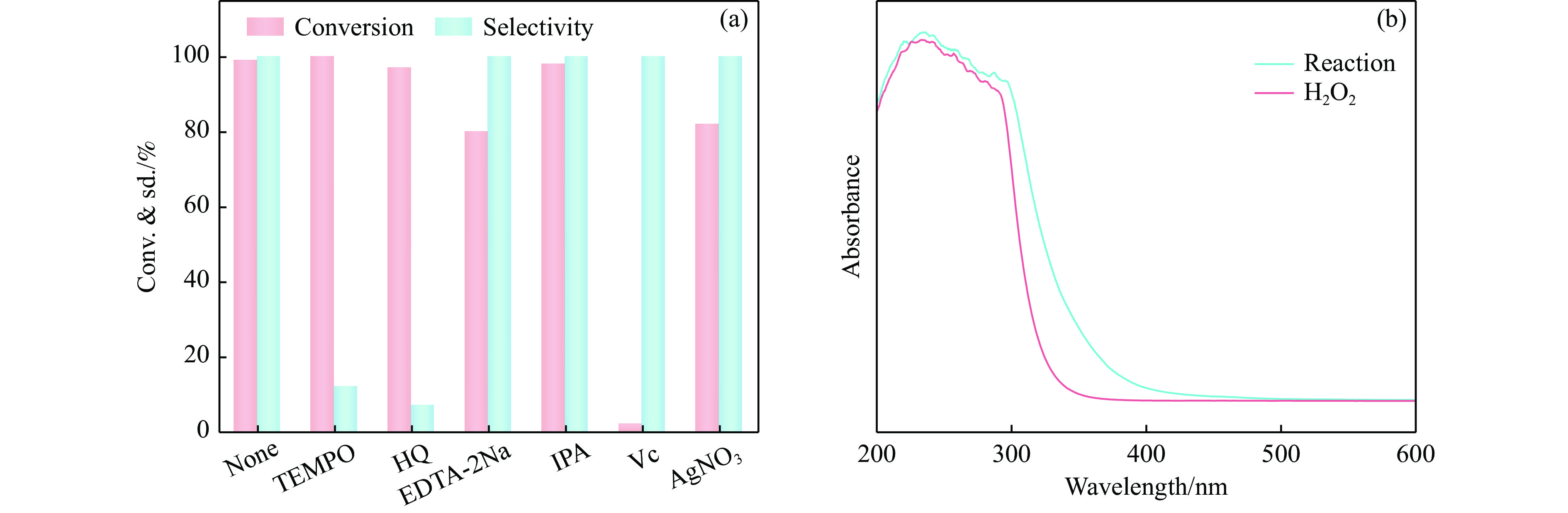
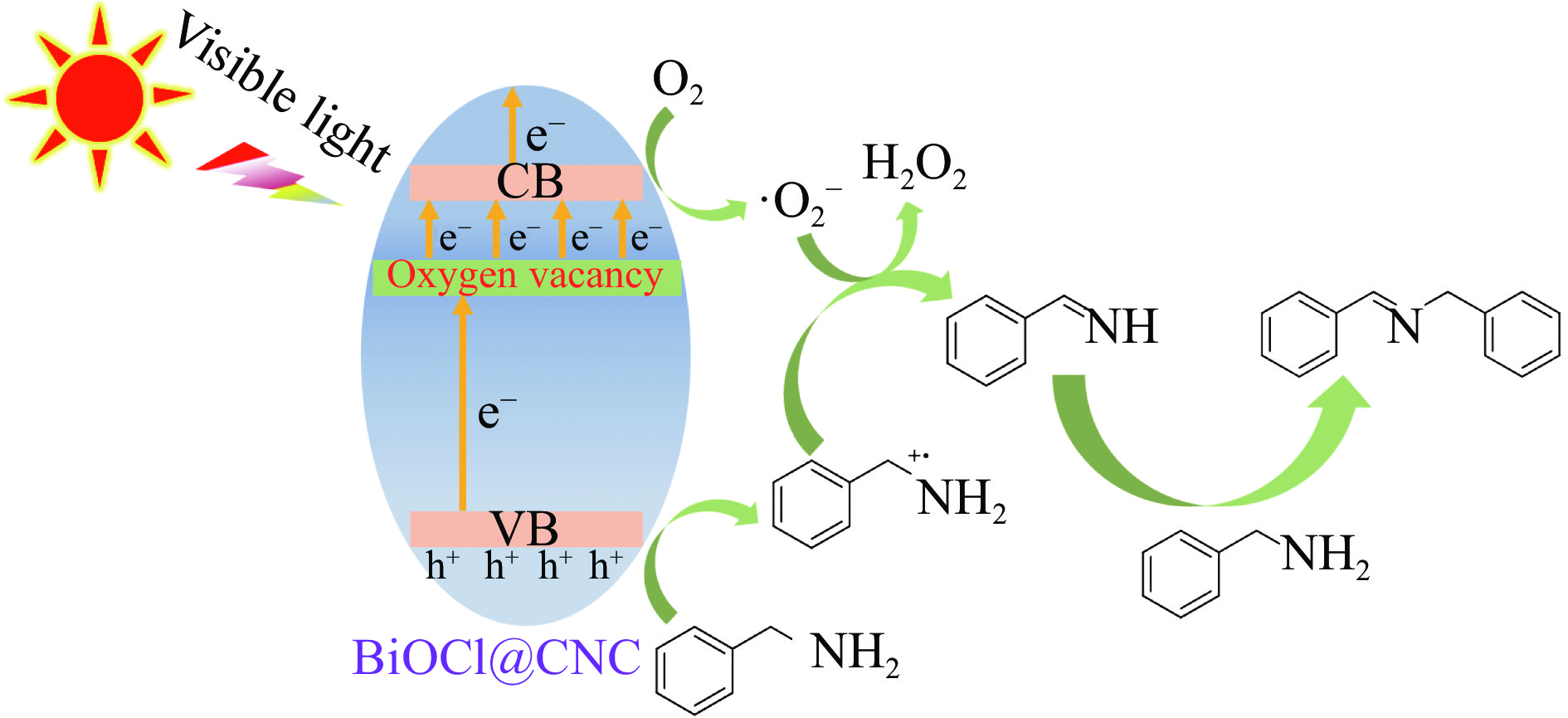
本工作采用纳米纤维素(CNC)为载体,与BiOCl在室温搅拌下制备了复合光催化剂BiOCl@CNC。XRD、FT-IR、SEM、TEM、XPS等系列表征表明,CNC中大量羟基可以与BiOCl通过氢键紧密结合,并在材料中构造丰富的氧空位,从而显著提升其可见光催化性能。以可见光下催化苄胺C−N偶联为目标反应评估BiOCl@CNC的性能并对机理进行研究。首先对反应条件进行优化,得最优条件为1.0 mmol苄胺作为底物,20 mg BiOCl@CNC作催化剂,以30 W 白色LED灯为光源,在CH3CN中氧气氛围下室温反应20 h。底物扩展实验表明,BiOCl@CNC对含有不同取代基的反应物均表现出良好的适应性,且具有优异的稳定性。通过自由基捕获实验表明,电子在氧空位的辅助下产生超氧自由基,并与胺阳离子自由基中间体形成最终产物。这一工作不仅丰富了Bi基复合半导体的应用,也为N-苄烯丁胺的合成提供了新思路。
本工作采用纳米纤维素(CNC)为载体,与BiOCl在室温搅拌下制备了复合光催化剂BiOCl@CNC。XRD、FT-IR、SEM、TEM、XPS等系列表征表明,CNC中大量羟基可以与BiOCl通过氢键紧密结合,并在材料中构造丰富的氧空位,从而显著提升其可见光催化性能。以可见光下催化苄胺C−N偶联为目标反应评估BiOCl@CNC的性能并对机理进行研究。首先对反应条件进行优化,得最优条件为1.0 mmol苄胺作为底物,20 mg BiOCl@CNC作催化剂,以30 W 白色LED灯为光源,在CH3CN中氧气氛围下室温反应20 h。底物扩展实验表明,BiOCl@CNC对含有不同取代基的反应物均表现出良好的适应性,且具有优异的稳定性。通过自由基捕获实验表明,电子在氧空位的辅助下产生超氧自由基,并与胺阳离子自由基中间体形成最终产物。这一工作不仅丰富了Bi基复合半导体的应用,也为N-苄烯丁胺的合成提供了新思路。


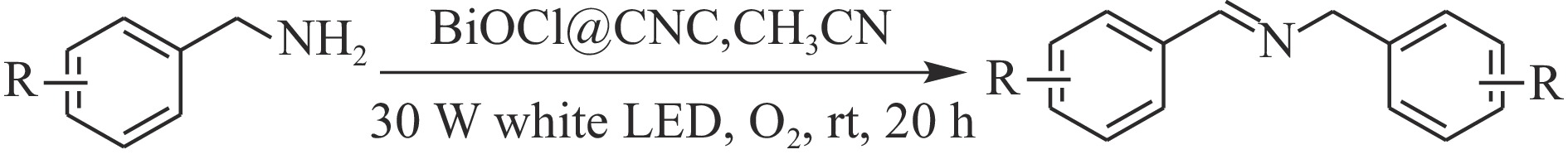
当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024013
摘要:
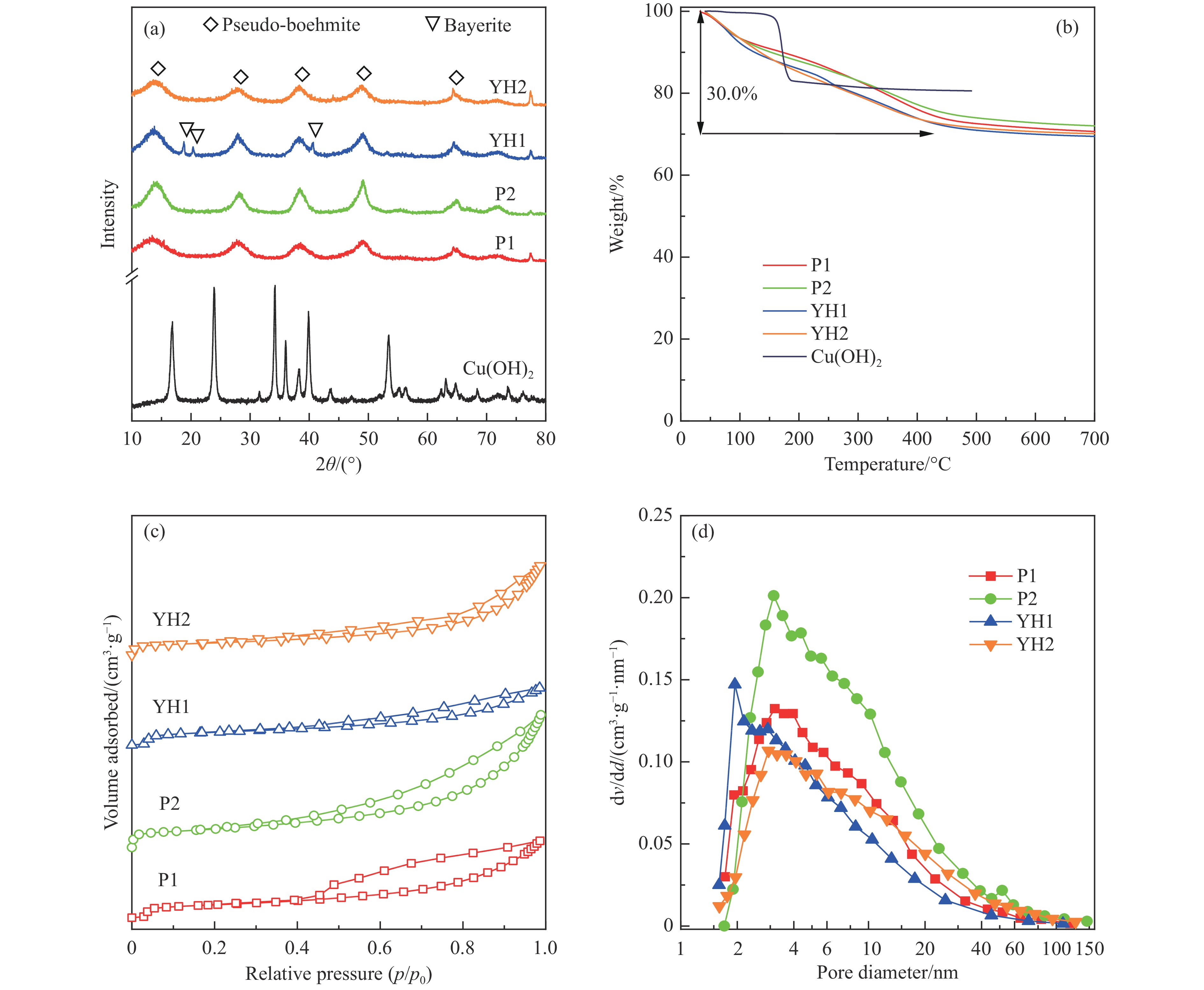
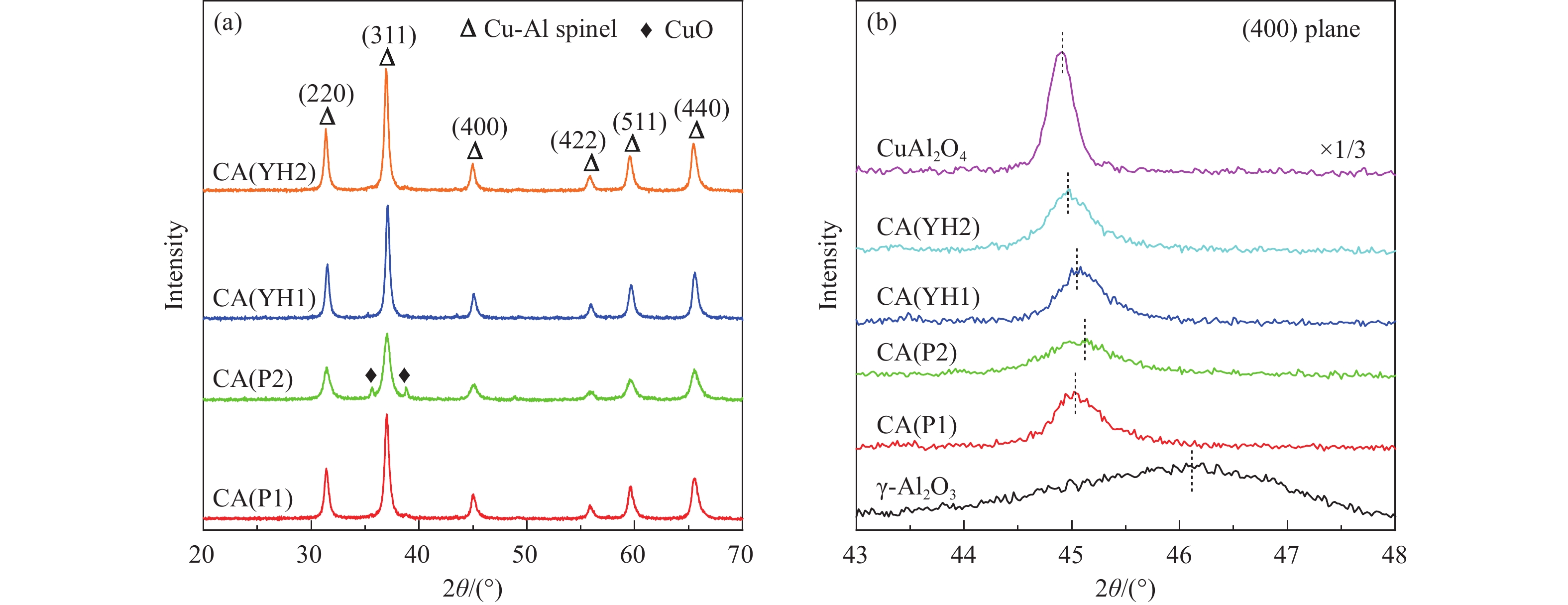
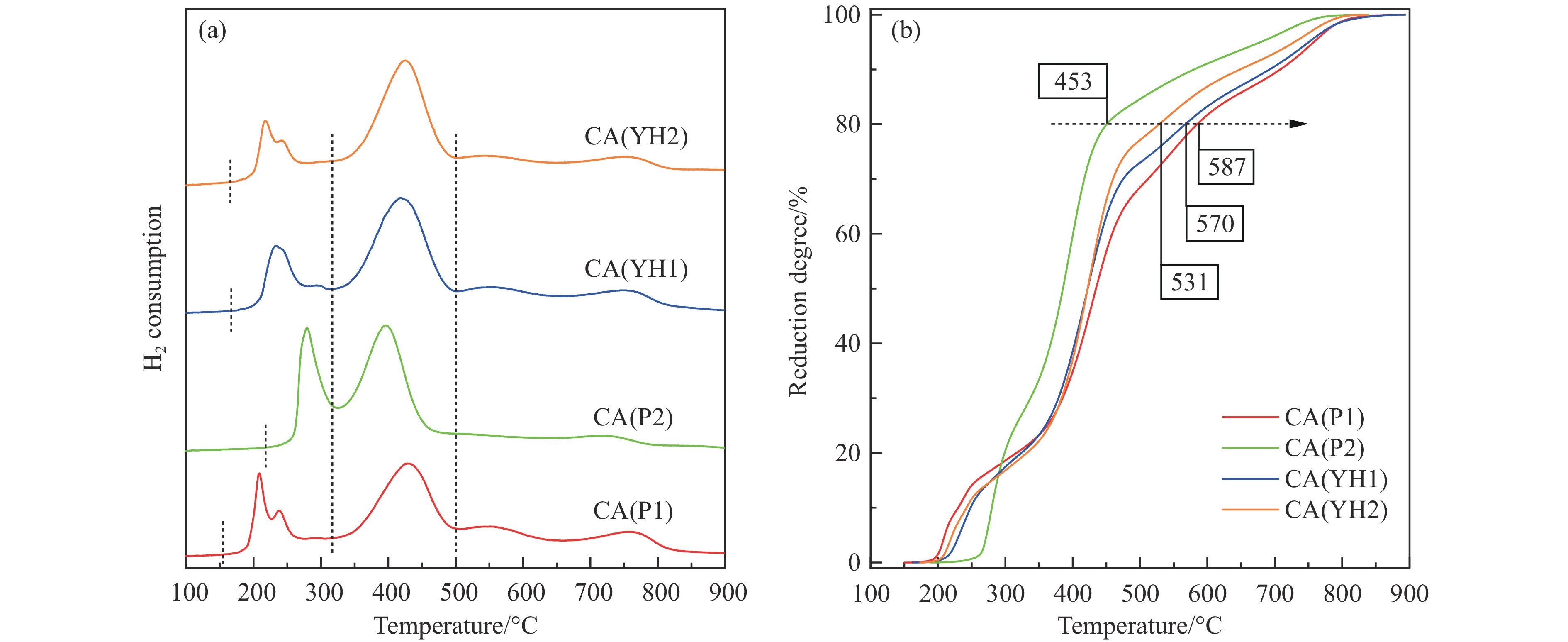
基于高能球磨和固相焙烧的方法,采用杂质元素(Na、Fe、Si和S)含量不等的四种拟薄水铝石和氢氧化铜制备了Cu-Al尖晶石固溶体催化剂,通过ICP-AES、TG、XRD、H2-TPR和BET表征了催化剂的物化性质,并考察了对逆水煤气变换反应的催化性能。结果表明,拟薄水铝石中的杂质元素对于Cu-Al尖晶石催化剂的物相性质、还原性能、织构性质和催化性能有显著的影响,Si有助于合成高比表面积的催化剂,但不利于Cu-Al尖晶石生成,导致催化活性低;含有少量Na和Fe的尖晶石的催化活性较低;S物种经高温焙烧后分解,对催化活性没有影响;基于杂质元素含量最低的拟薄水铝石合成的催化剂中难还原尖晶石含量最高,表现出最高的逆水煤气变换催化活性。此外,基于活性最优样品的CO2-TPD-MS和In-situ DRIFTS分析表明,Al上形成的双齿甲酸盐是Cu-Al尖晶石固溶体催化CO2加氢生成CO的主要中间产物,其含量与催化活性随反应时间的变化规律一致。
基于高能球磨和固相焙烧的方法,采用杂质元素(Na、Fe、Si和S)含量不等的四种拟薄水铝石和氢氧化铜制备了Cu-Al尖晶石固溶体催化剂,通过ICP-AES、TG、XRD、H2-TPR和BET表征了催化剂的物化性质,并考察了对逆水煤气变换反应的催化性能。结果表明,拟薄水铝石中的杂质元素对于Cu-Al尖晶石催化剂的物相性质、还原性能、织构性质和催化性能有显著的影响,Si有助于合成高比表面积的催化剂,但不利于Cu-Al尖晶石生成,导致催化活性低;含有少量Na和Fe的尖晶石的催化活性较低;S物种经高温焙烧后分解,对催化活性没有影响;基于杂质元素含量最低的拟薄水铝石合成的催化剂中难还原尖晶石含量最高,表现出最高的逆水煤气变换催化活性。此外,基于活性最优样品的CO2-TPD-MS和In-situ DRIFTS分析表明,Al上形成的双齿甲酸盐是Cu-Al尖晶石固溶体催化CO2加氢生成CO的主要中间产物,其含量与催化活性随反应时间的变化规律一致。

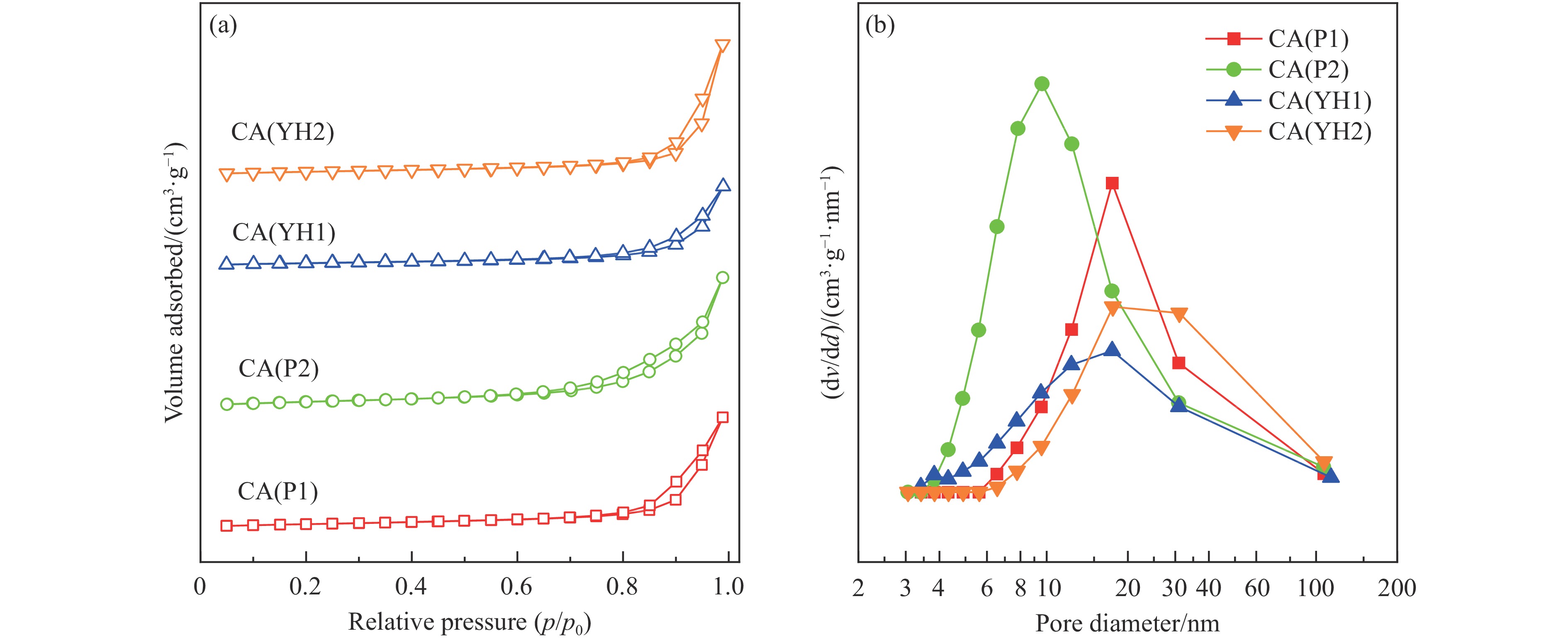
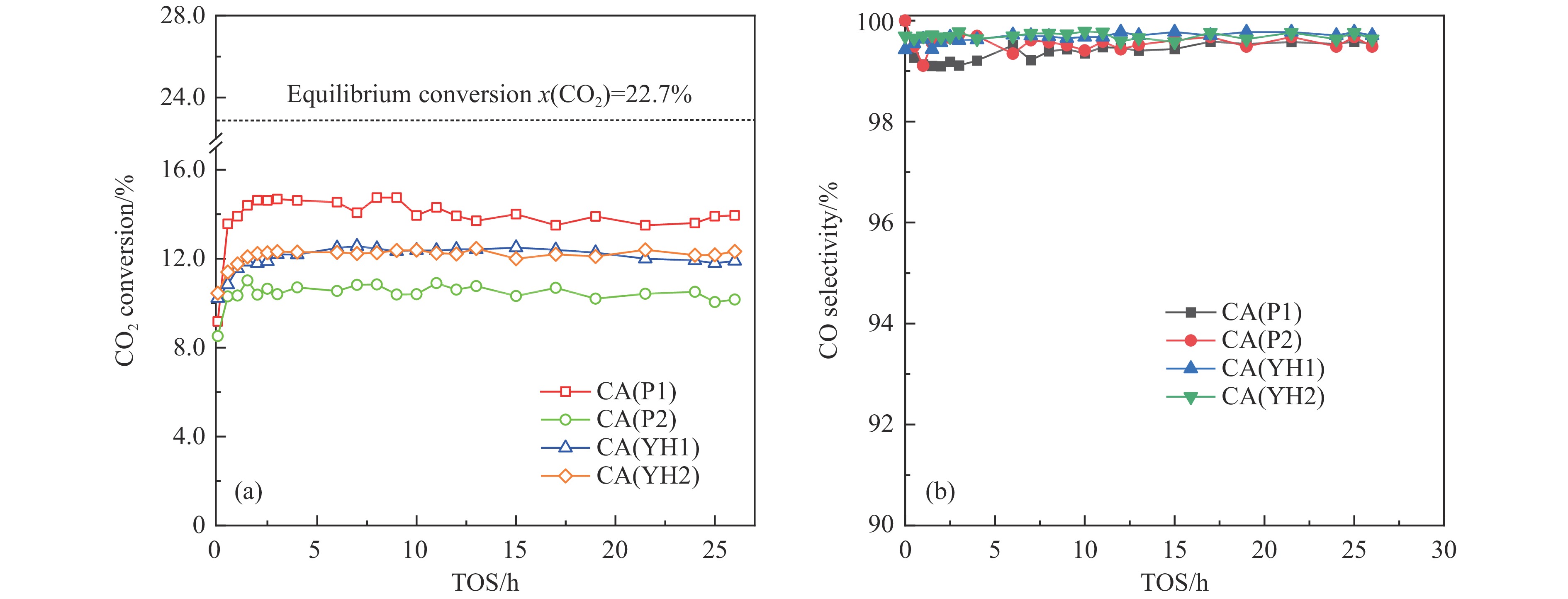
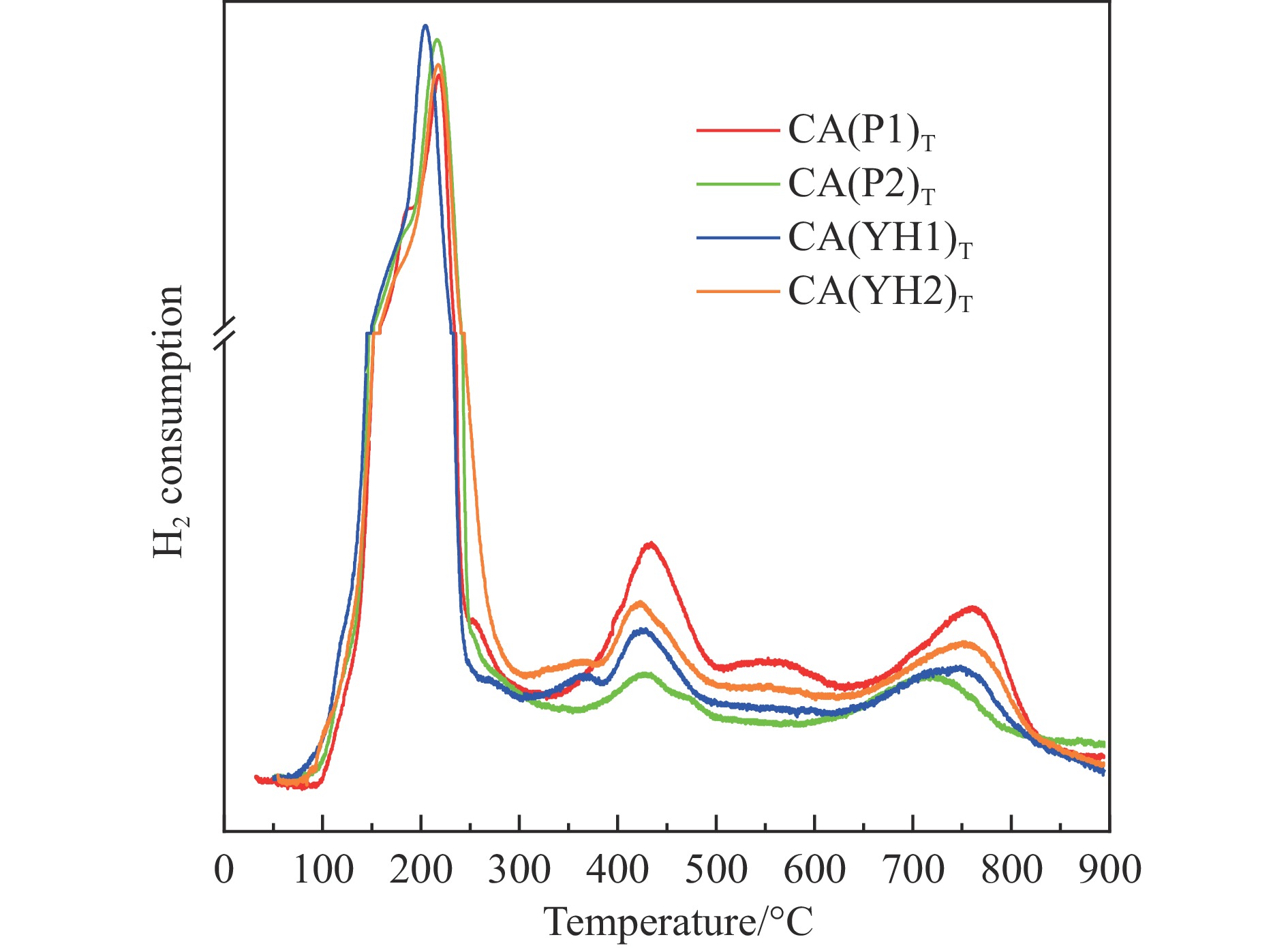
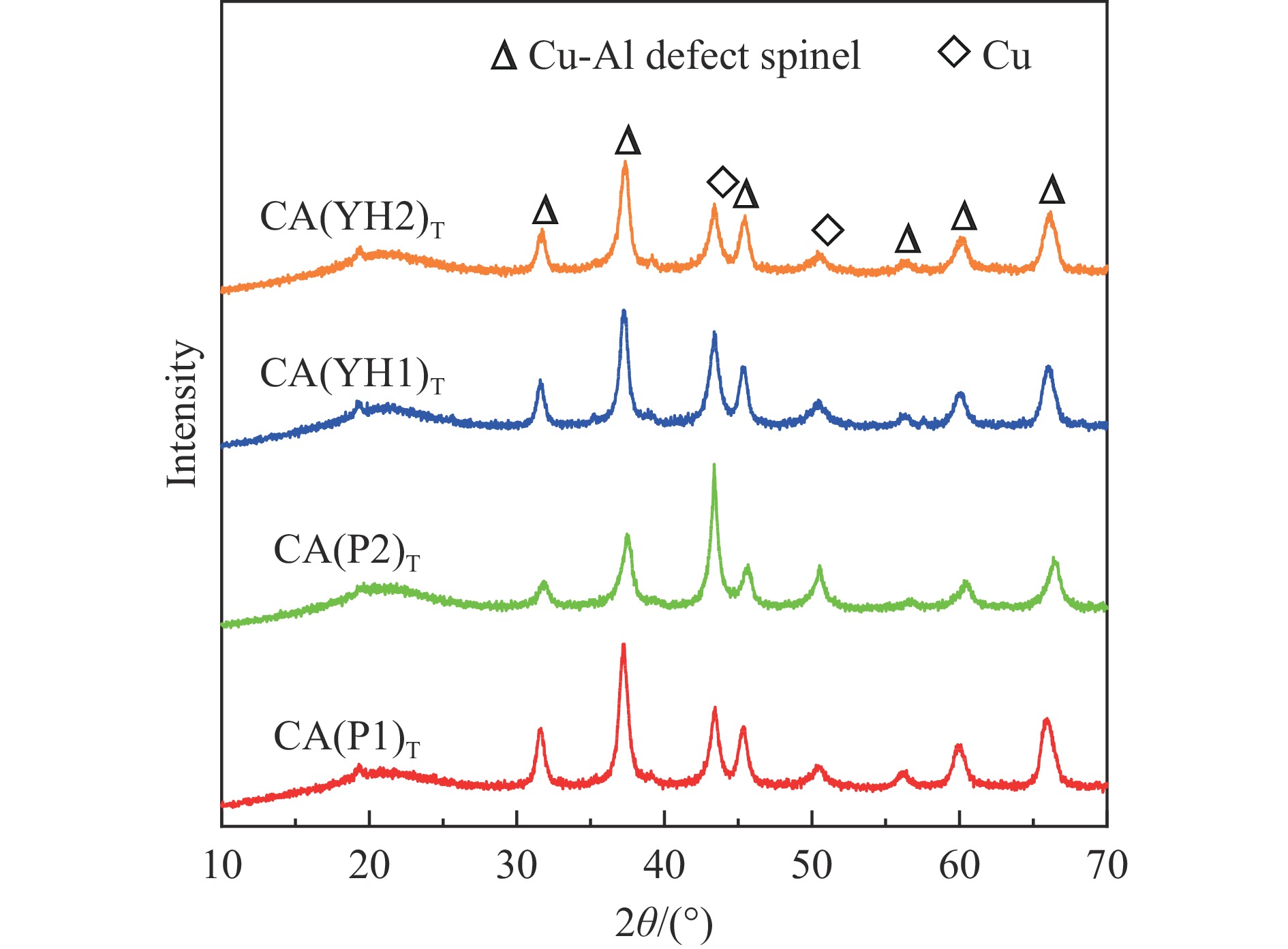
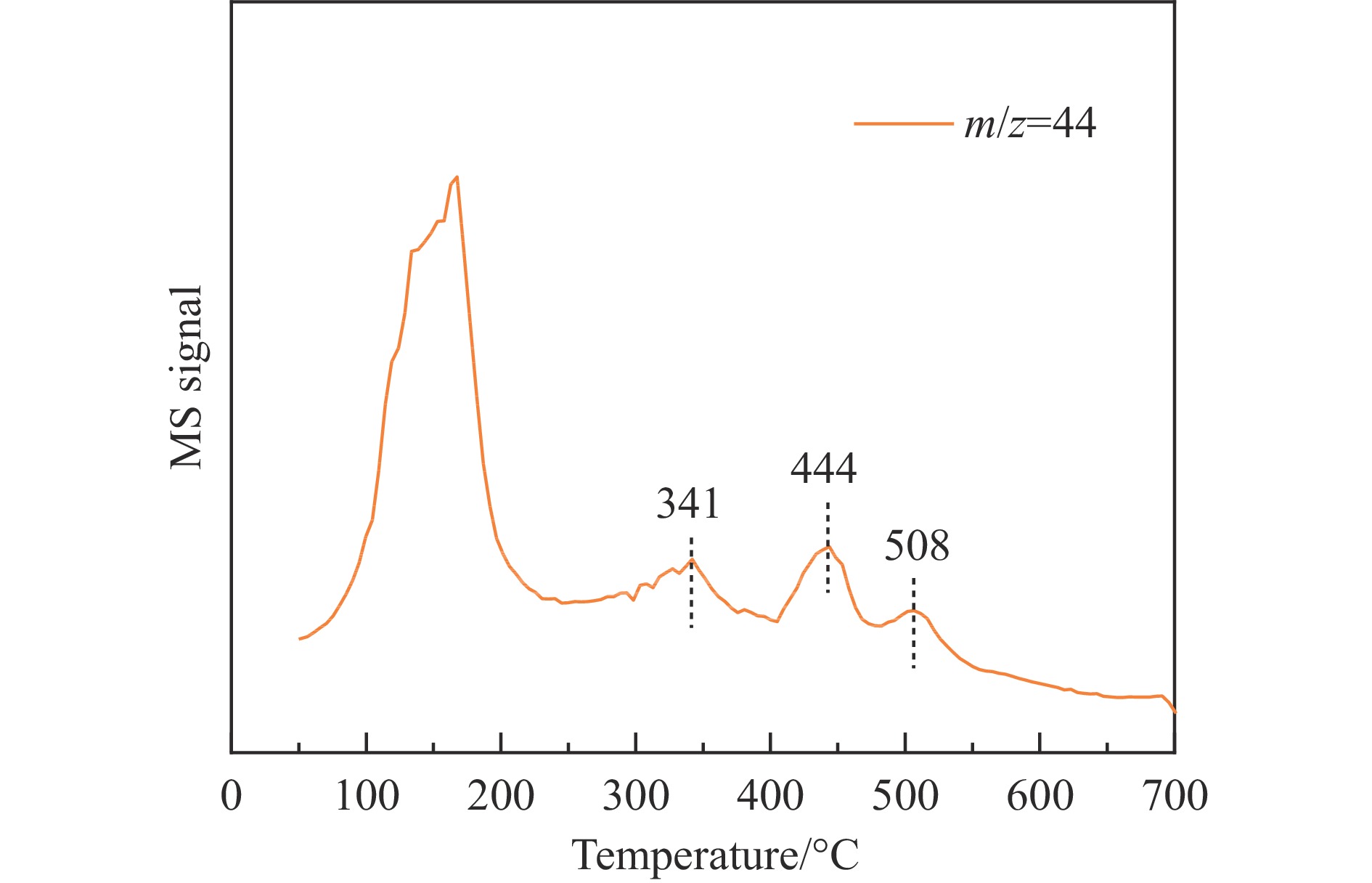


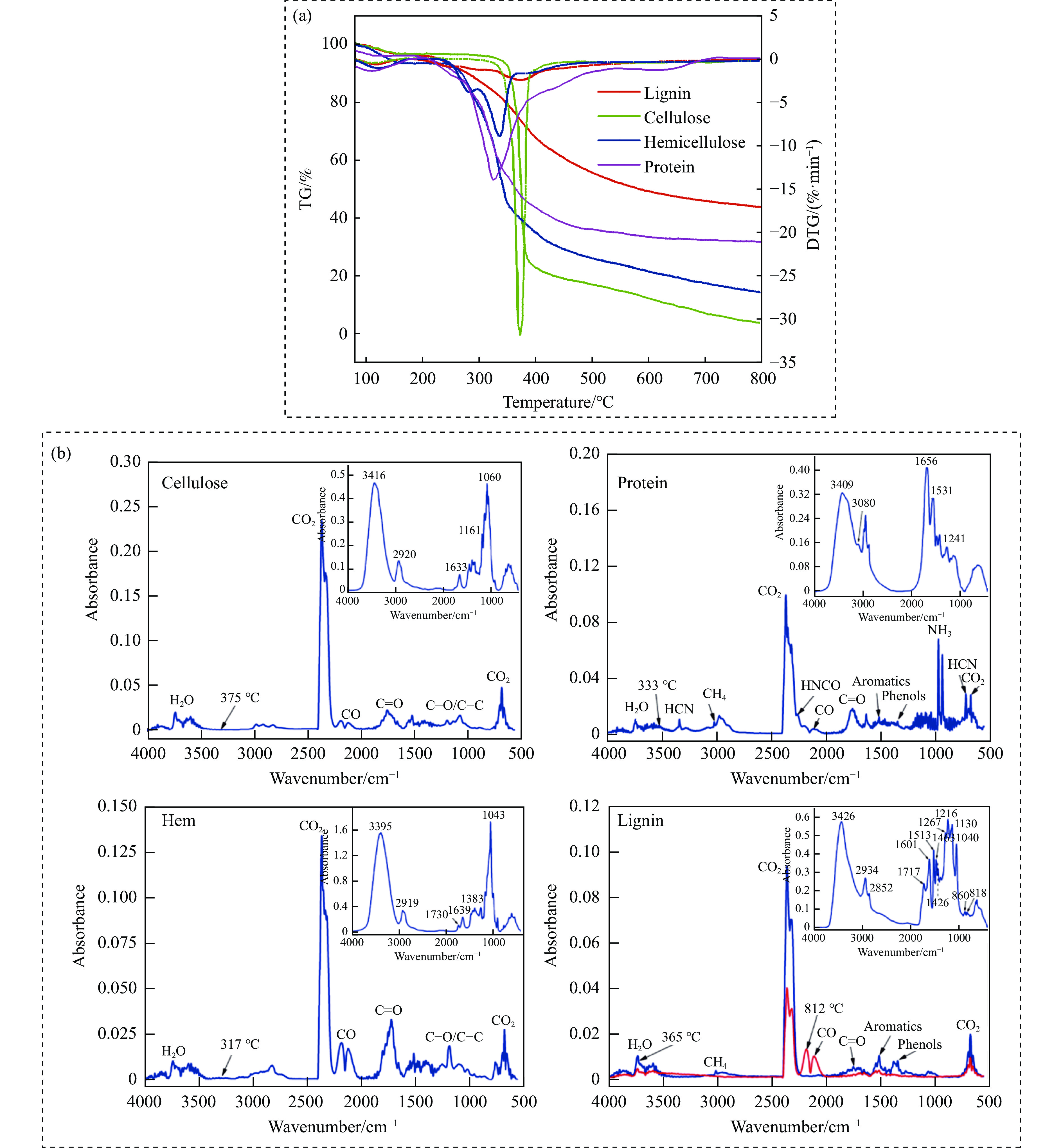
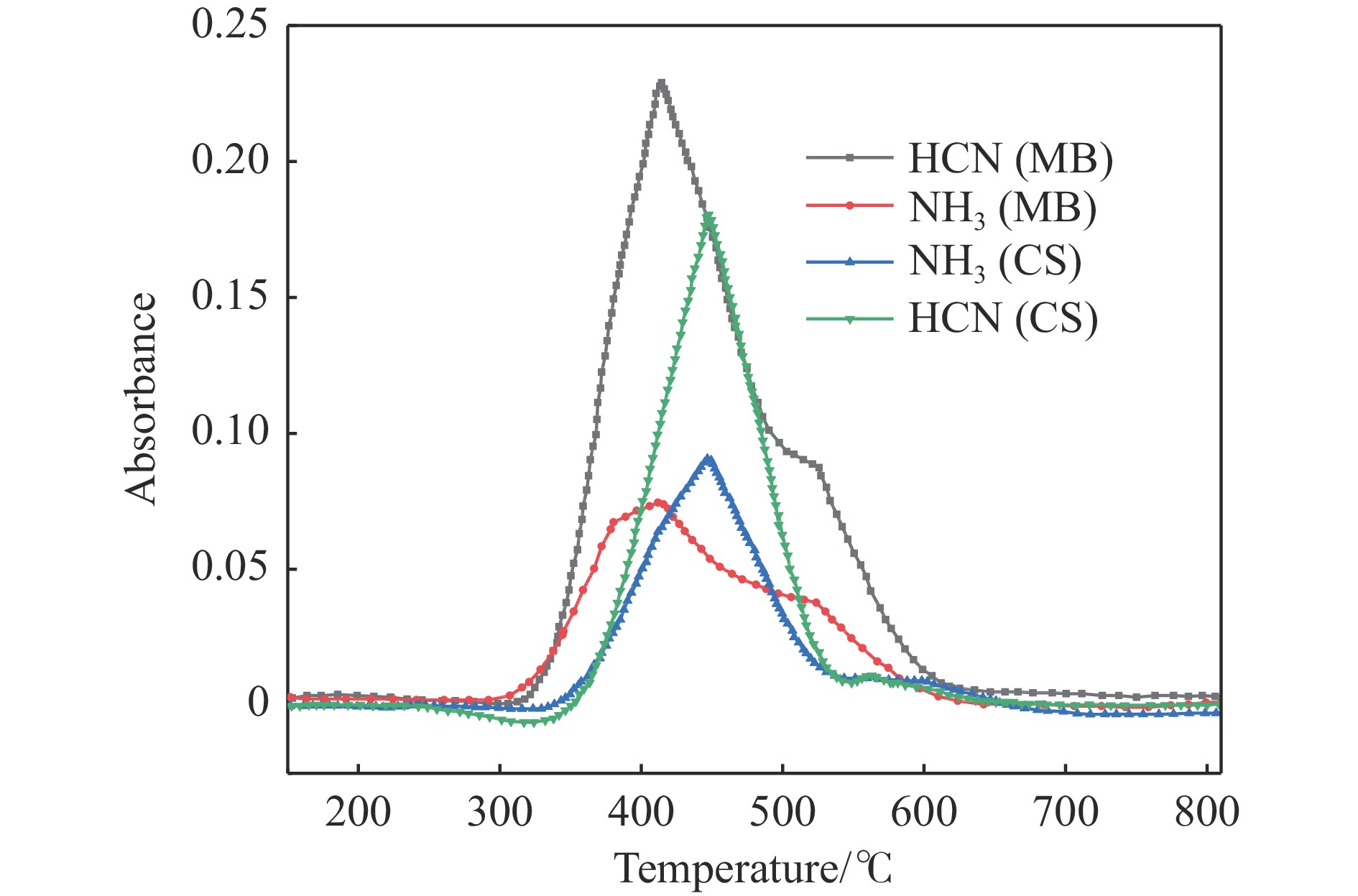
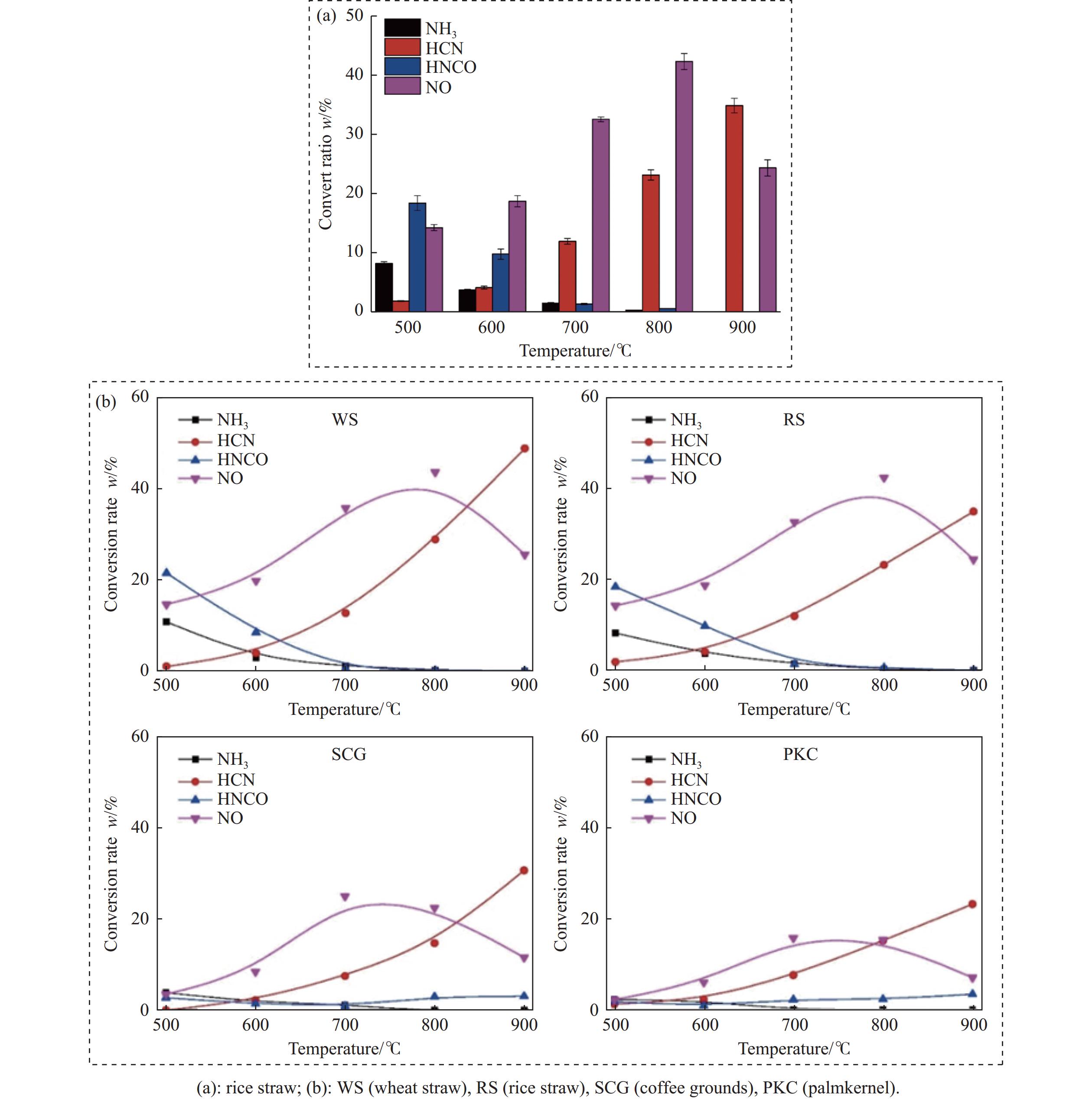
当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023090
摘要:
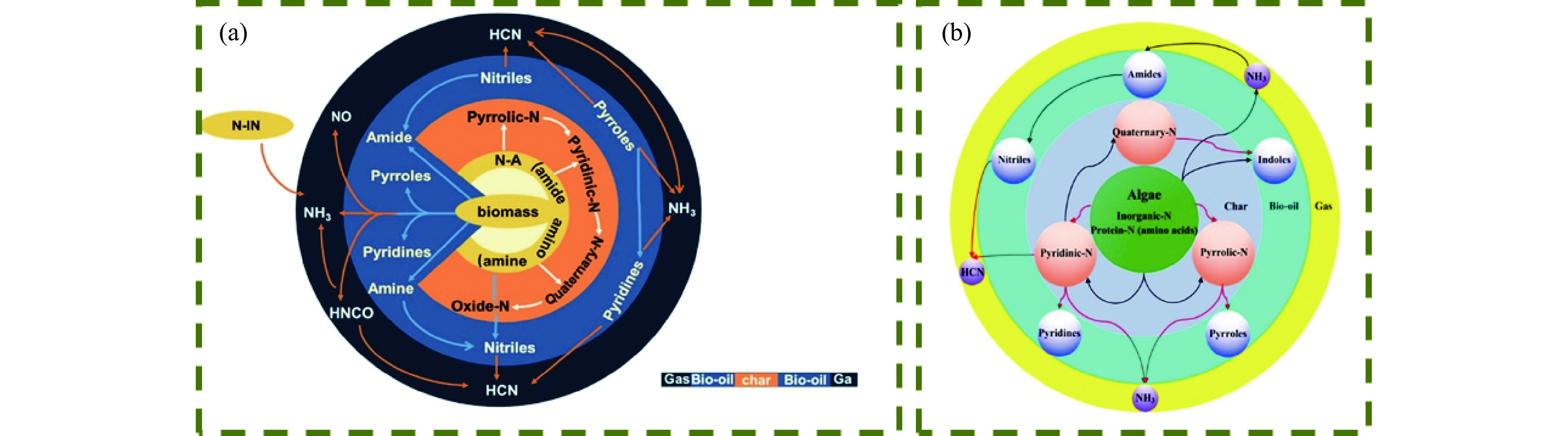
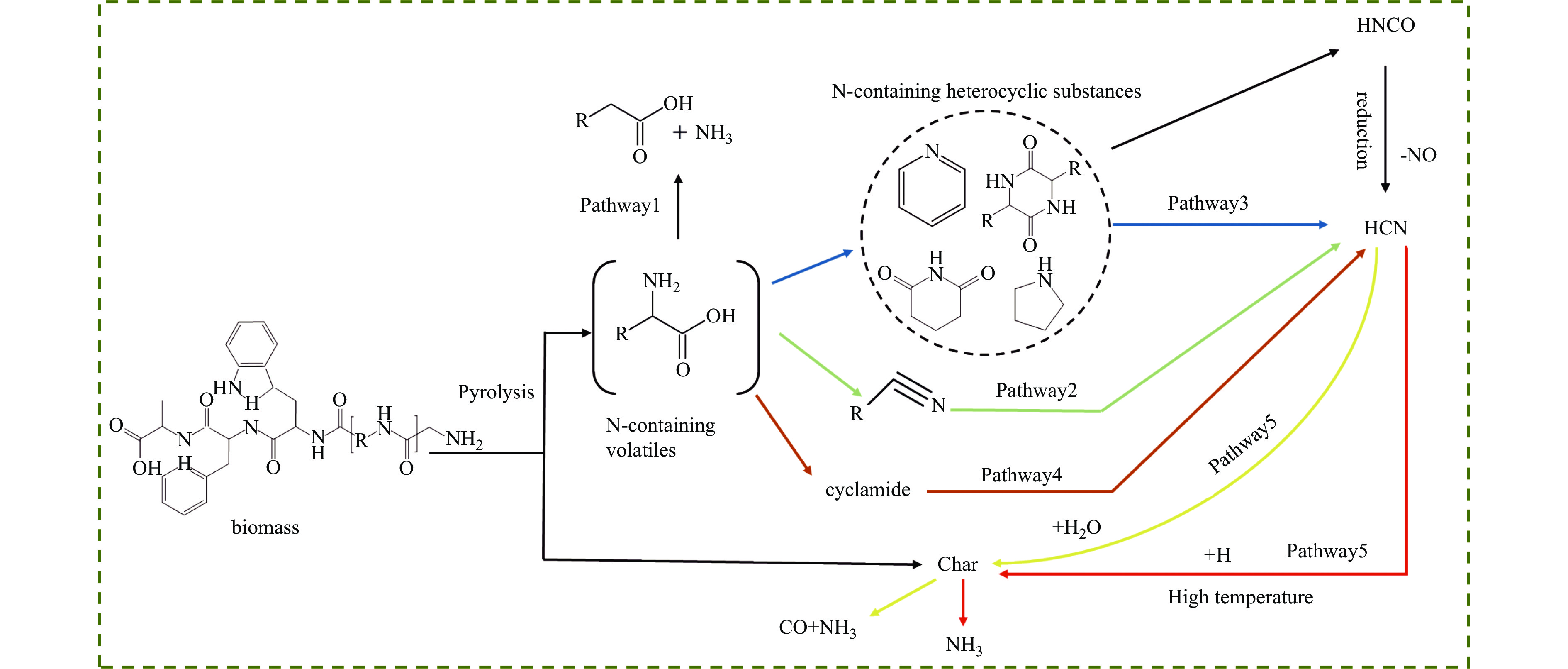
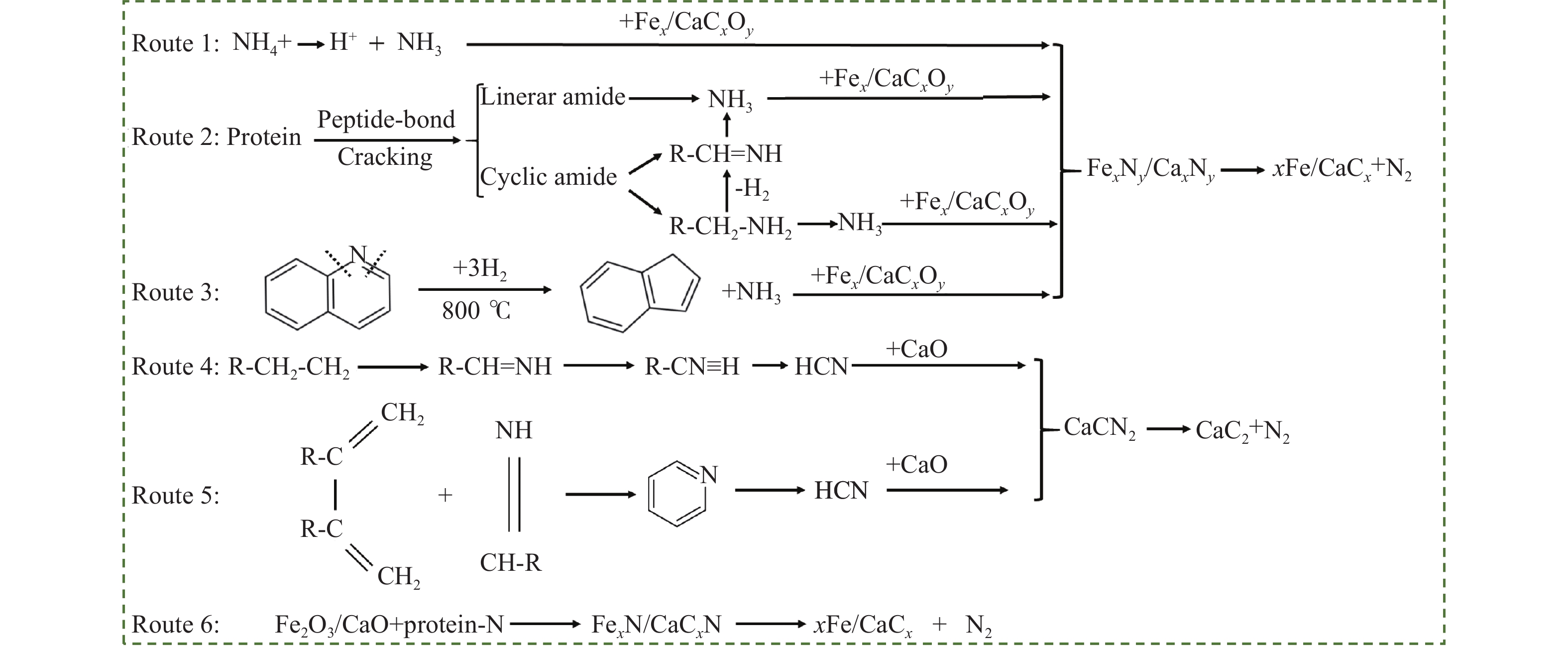
热解是利用生物质能的一种高效且经济的方式,但生物质热解气中的含氮化合物使热解气品质低且燃烧导致空气二次污染。本工作总结了生物质热解气中的含氮化合物研究现状,主要综述了典型生物质热失重行为,探讨了生物质热解气中含氮化合物的生成机理,分析了含氮化合物的分布状况和控制的研究进展。同时,指出了含氮化合物控制在实际应用中面临的困难挑战,进一步展望了含氮化合物控制工艺优化及经济性分析的重点研究方向,为生物质热解气净化提供理论依据和技术支持。
热解是利用生物质能的一种高效且经济的方式,但生物质热解气中的含氮化合物使热解气品质低且燃烧导致空气二次污染。本工作总结了生物质热解气中的含氮化合物研究现状,主要综述了典型生物质热失重行为,探讨了生物质热解气中含氮化合物的生成机理,分析了含氮化合物的分布状况和控制的研究进展。同时,指出了含氮化合物控制在实际应用中面临的困难挑战,进一步展望了含氮化合物控制工艺优化及经济性分析的重点研究方向,为生物质热解气净化提供理论依据和技术支持。


当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60432-9
摘要:
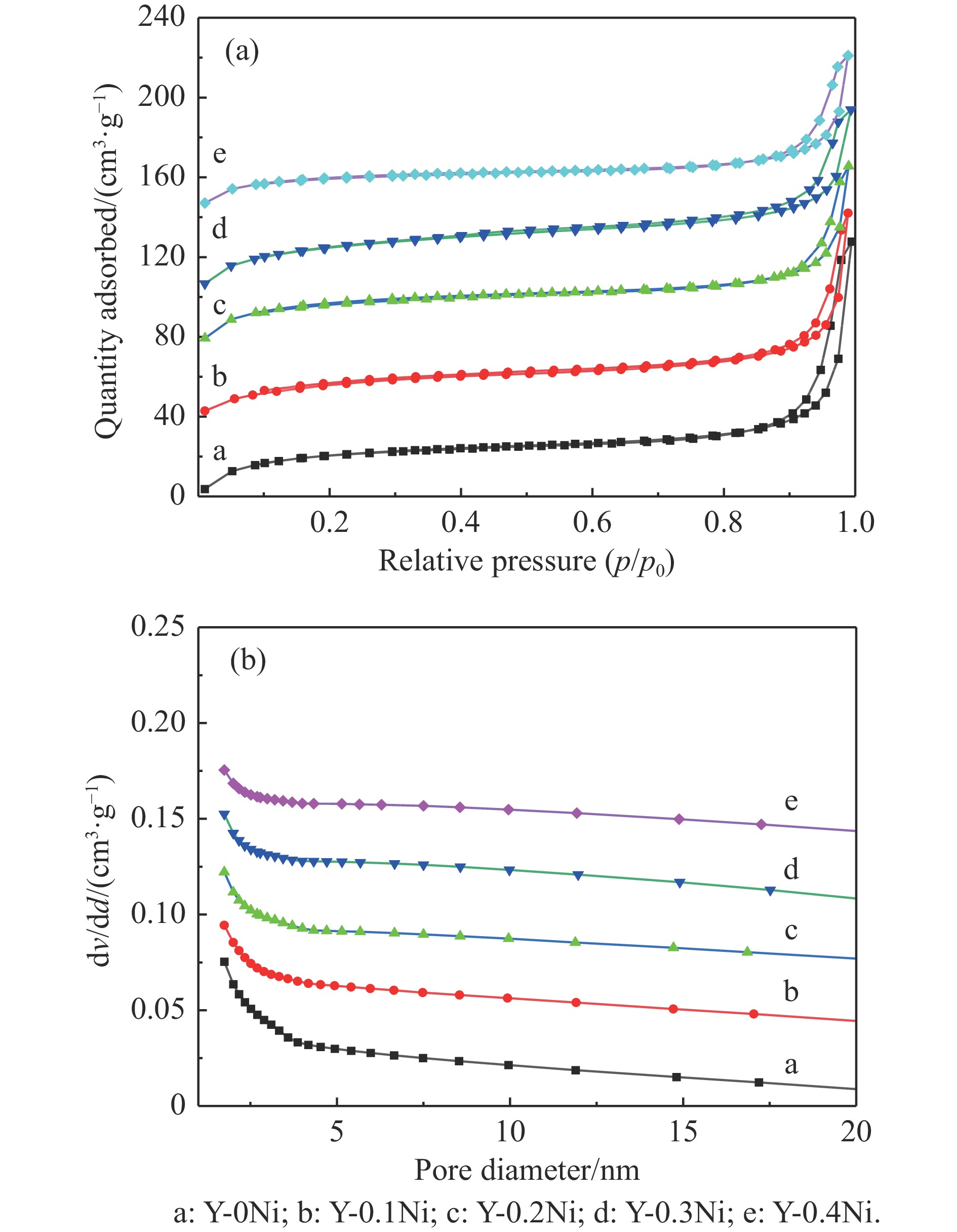
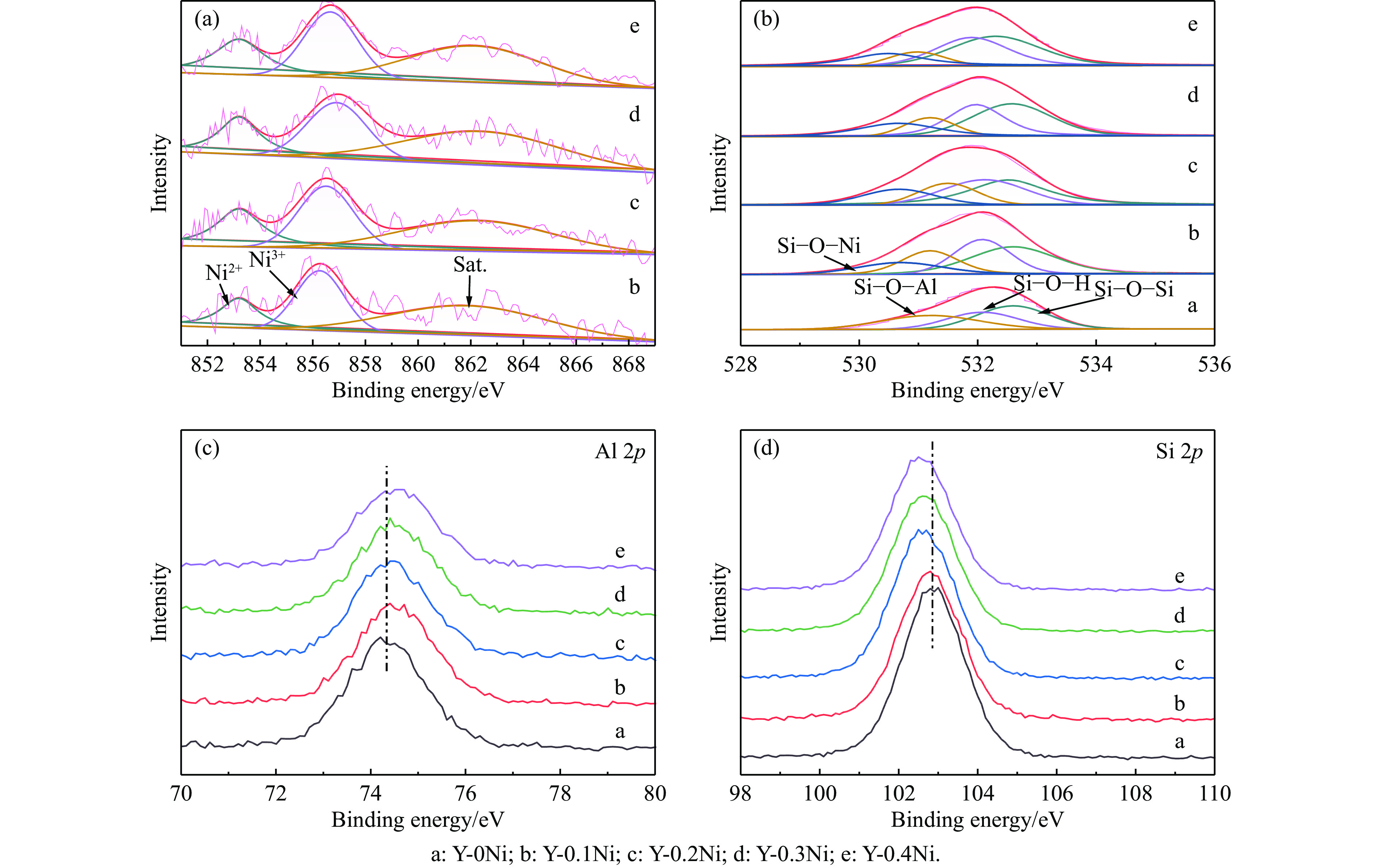
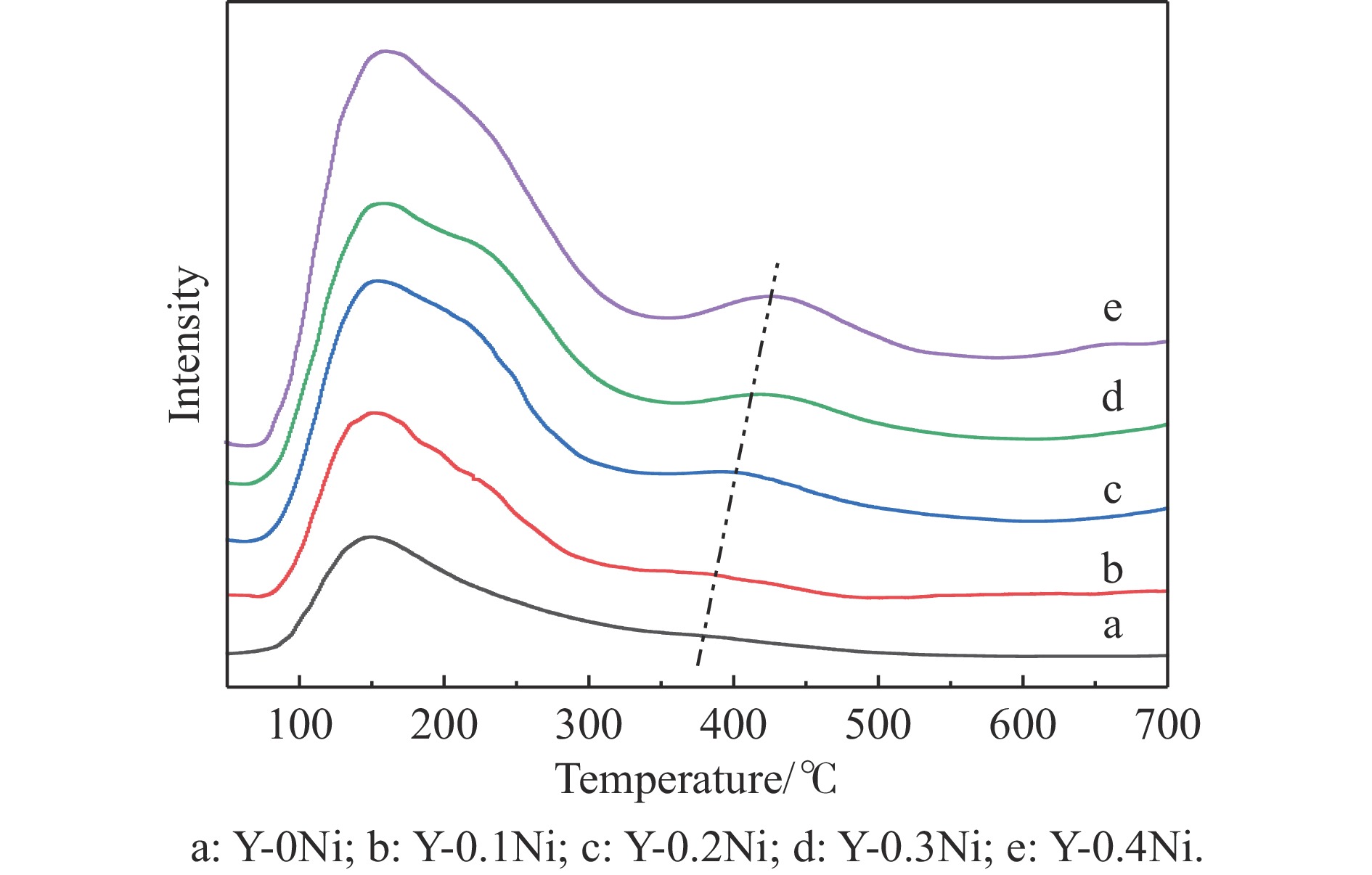
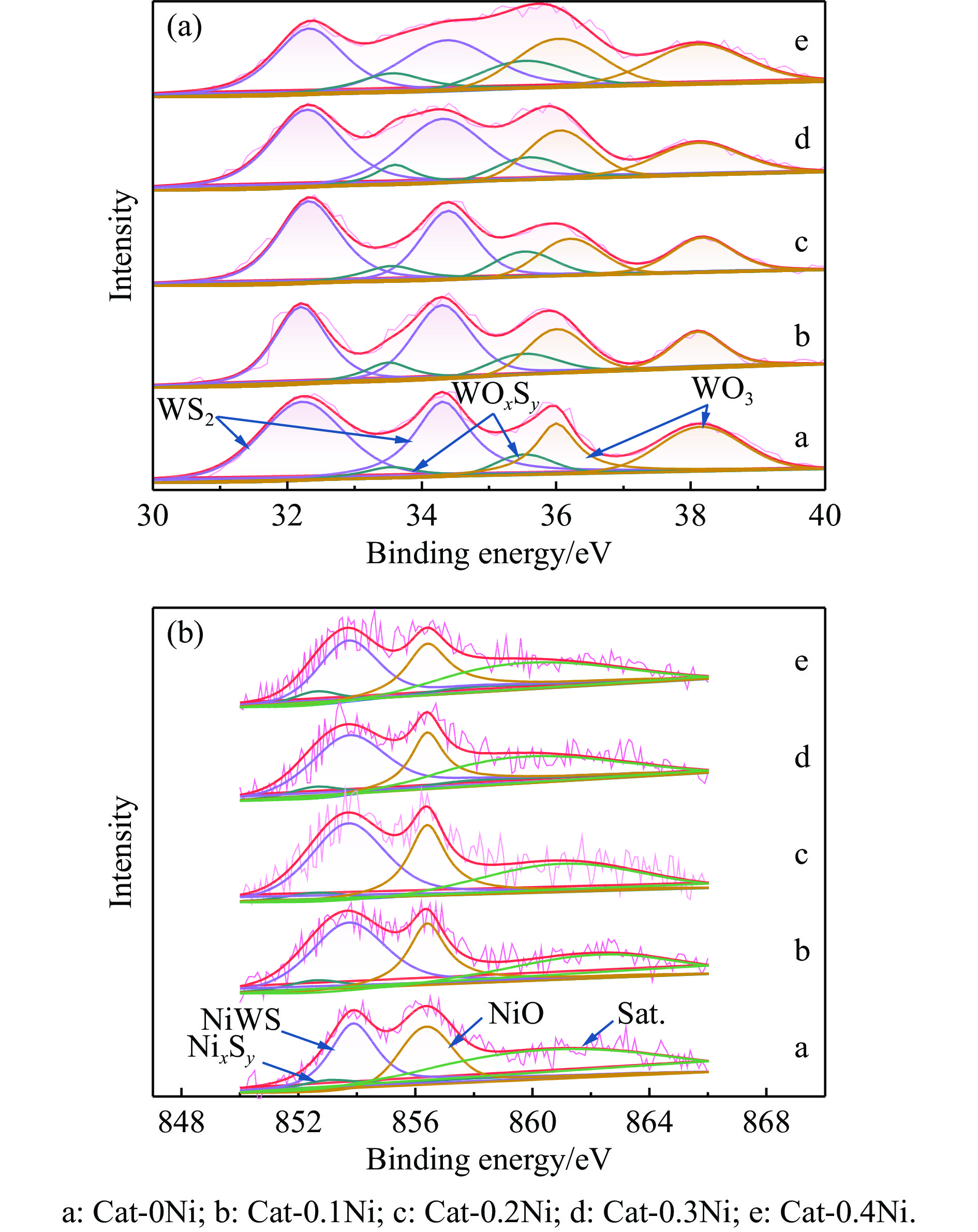
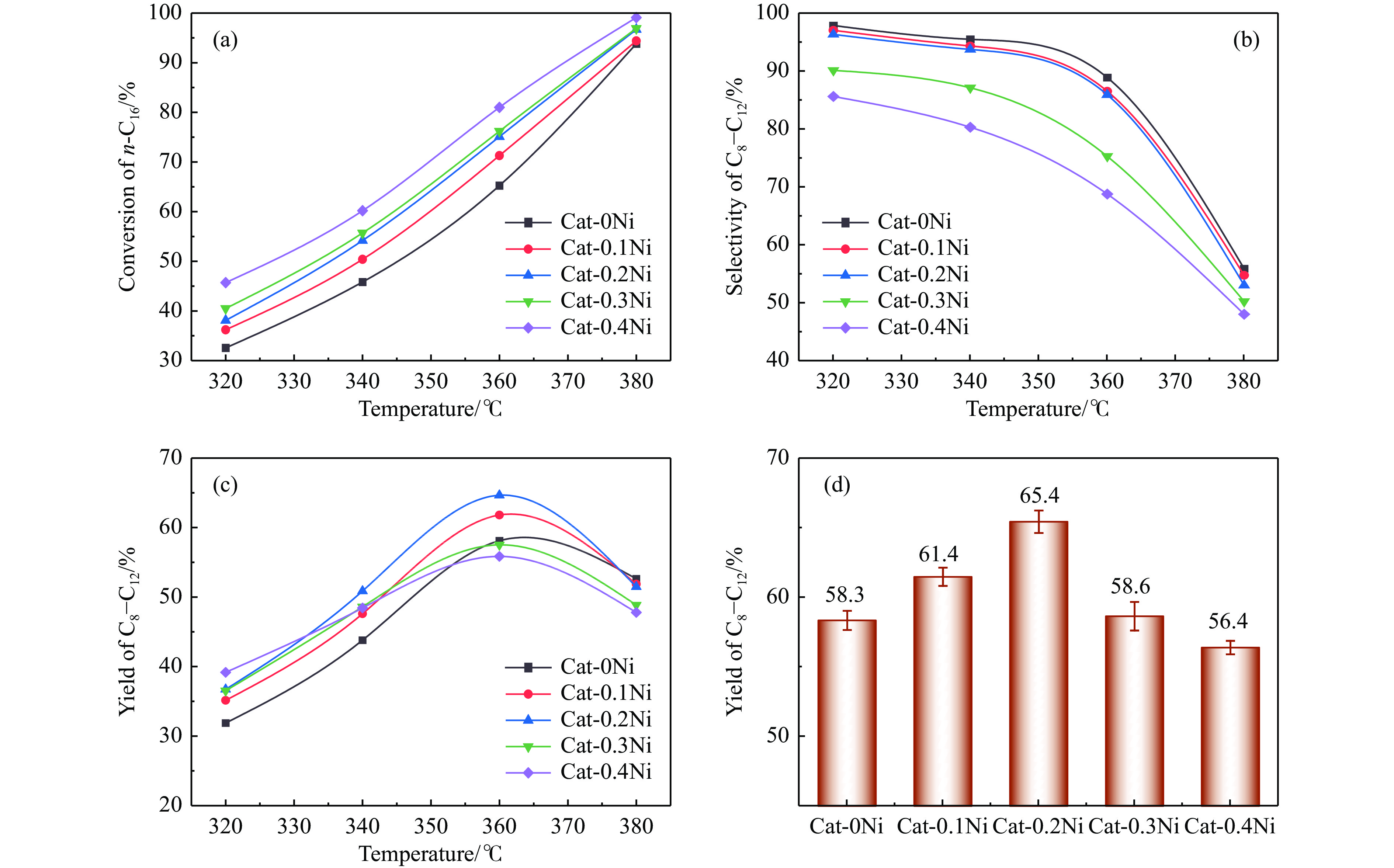
通过原位合成法在小晶粒Y分子筛合成的过程中,引入Ni源合成了一系列不同Ni掺入量的小晶粒Y-xNi分子筛,将活性金属Ni预浸渍到Y分子筛的骨架中。将Y-xNi分子筛和ASA混合作为载体并采用等体积浸渍法负载Ni、W制备Cat-xNi系列加氢裂化催化剂。以正十六烷为反应物,探究其加氢裂化反应性能。采用扫描电子显微镜(SEM)、X-射线衍射(XRD)、N2吸附-脱附、氨气程序升温脱附(NH3-TPD)、氢气程序升温还原(H2-TPR)、透射电子显微镜(TEM)和X射线光电子能谱(XPS)等表征手段分析了Ni的掺入对Y分子筛及催化剂理化性质的影响。结果表明,Ni主要取代Al引入Y分子筛骨架。在Y分子筛中适量掺入Ni会提高Y分子筛的相对结晶度以及Brønsted酸和Lewis酸位点的数量,但过量的Ni掺入不利于Y分子筛的结晶。Ni掺入削弱了金属与载体间的相互作用,提高了活性金属的硫化度及NiWS活性相的堆积数及分散度,调节了催化剂上金属中心与酸中心的匹配。催化性能评价表明,Ni改性有利于提高中间馏分产物(C8−C12)的选择性及收率。即同时增加Brønsted酸中心与NiWS活性中心数量、提高了金属中心与酸中心之间的协同作用,在提高转化率的同时避免过度裂化,提高中间馏分产物的收率。在360 ℃反应温度下,催化剂Cat-0.2Ni具有较高的n-C16转化率和C8−C12产物收率,n-C16转化率较Cat-0Ni提高了10.2个百分点,C8−C12产物收率为65.4%。采用原位合成法将活性金属Ni预浸渍在Y分子筛上可以有效调节裂化活性中心与加氢活性中心之间的平衡而提高催化活性和中间馏分产物的收率。
通过原位合成法在小晶粒Y分子筛合成的过程中,引入Ni源合成了一系列不同Ni掺入量的小晶粒Y-xNi分子筛,将活性金属Ni预浸渍到Y分子筛的骨架中。将Y-xNi分子筛和ASA混合作为载体并采用等体积浸渍法负载Ni、W制备Cat-xNi系列加氢裂化催化剂。以正十六烷为反应物,探究其加氢裂化反应性能。采用扫描电子显微镜(SEM)、X-射线衍射(XRD)、N2吸附-脱附、氨气程序升温脱附(NH3-TPD)、氢气程序升温还原(H2-TPR)、透射电子显微镜(TEM)和X射线光电子能谱(XPS)等表征手段分析了Ni的掺入对Y分子筛及催化剂理化性质的影响。结果表明,Ni主要取代Al引入Y分子筛骨架。在Y分子筛中适量掺入Ni会提高Y分子筛的相对结晶度以及Brønsted酸和Lewis酸位点的数量,但过量的Ni掺入不利于Y分子筛的结晶。Ni掺入削弱了金属与载体间的相互作用,提高了活性金属的硫化度及NiWS活性相的堆积数及分散度,调节了催化剂上金属中心与酸中心的匹配。催化性能评价表明,Ni改性有利于提高中间馏分产物(C8−C12)的选择性及收率。即同时增加Brønsted酸中心与NiWS活性中心数量、提高了金属中心与酸中心之间的协同作用,在提高转化率的同时避免过度裂化,提高中间馏分产物的收率。在360 ℃反应温度下,催化剂Cat-0.2Ni具有较高的n-C16转化率和C8−C12产物收率,n-C16转化率较Cat-0Ni提高了10.2个百分点,C8−C12产物收率为65.4%。采用原位合成法将活性金属Ni预浸渍在Y分子筛上可以有效调节裂化活性中心与加氢活性中心之间的平衡而提高催化活性和中间馏分产物的收率。


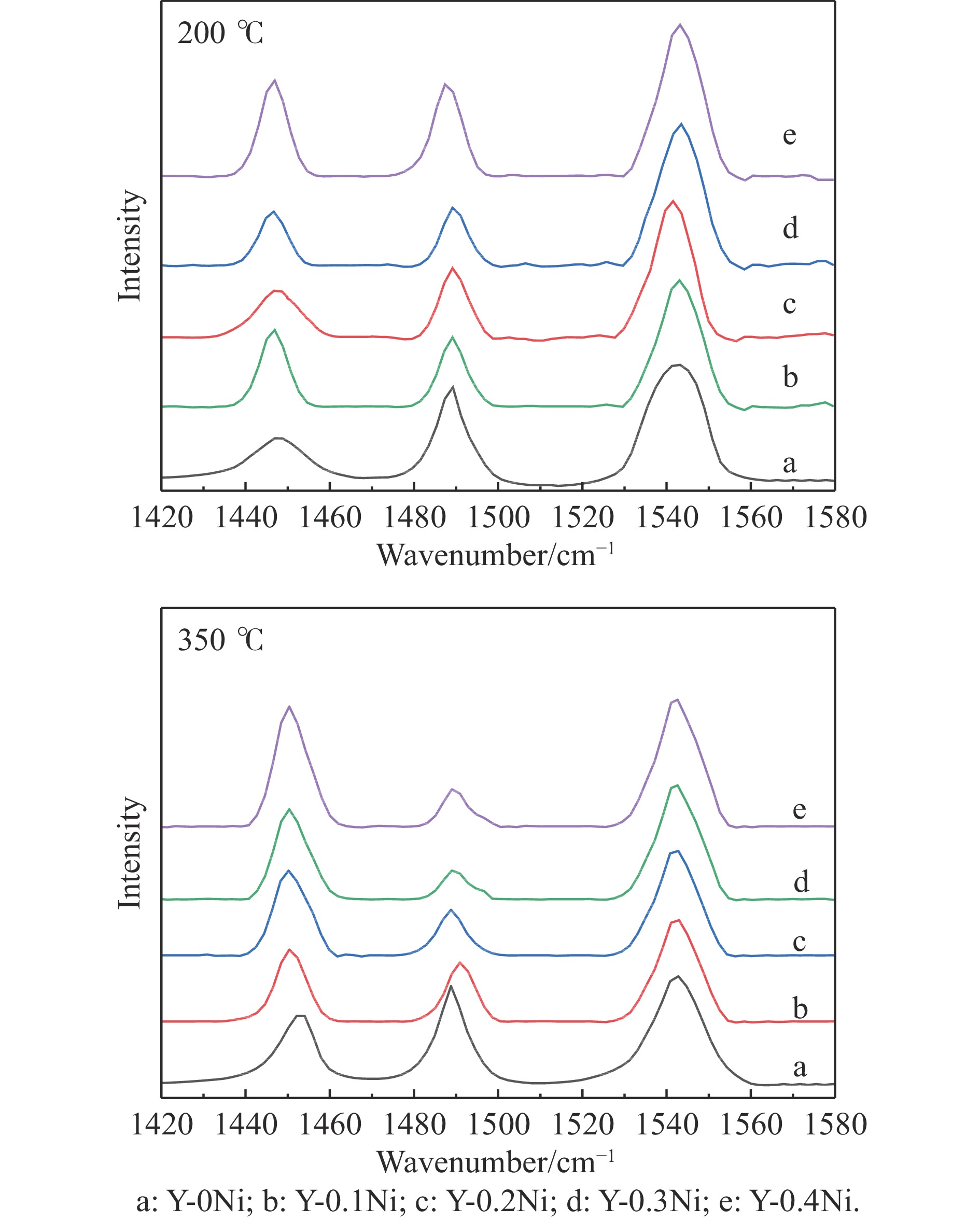
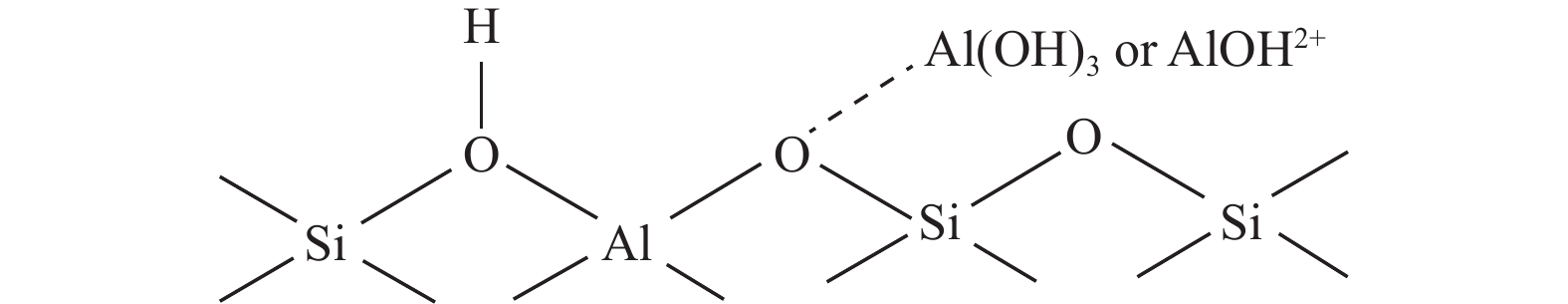
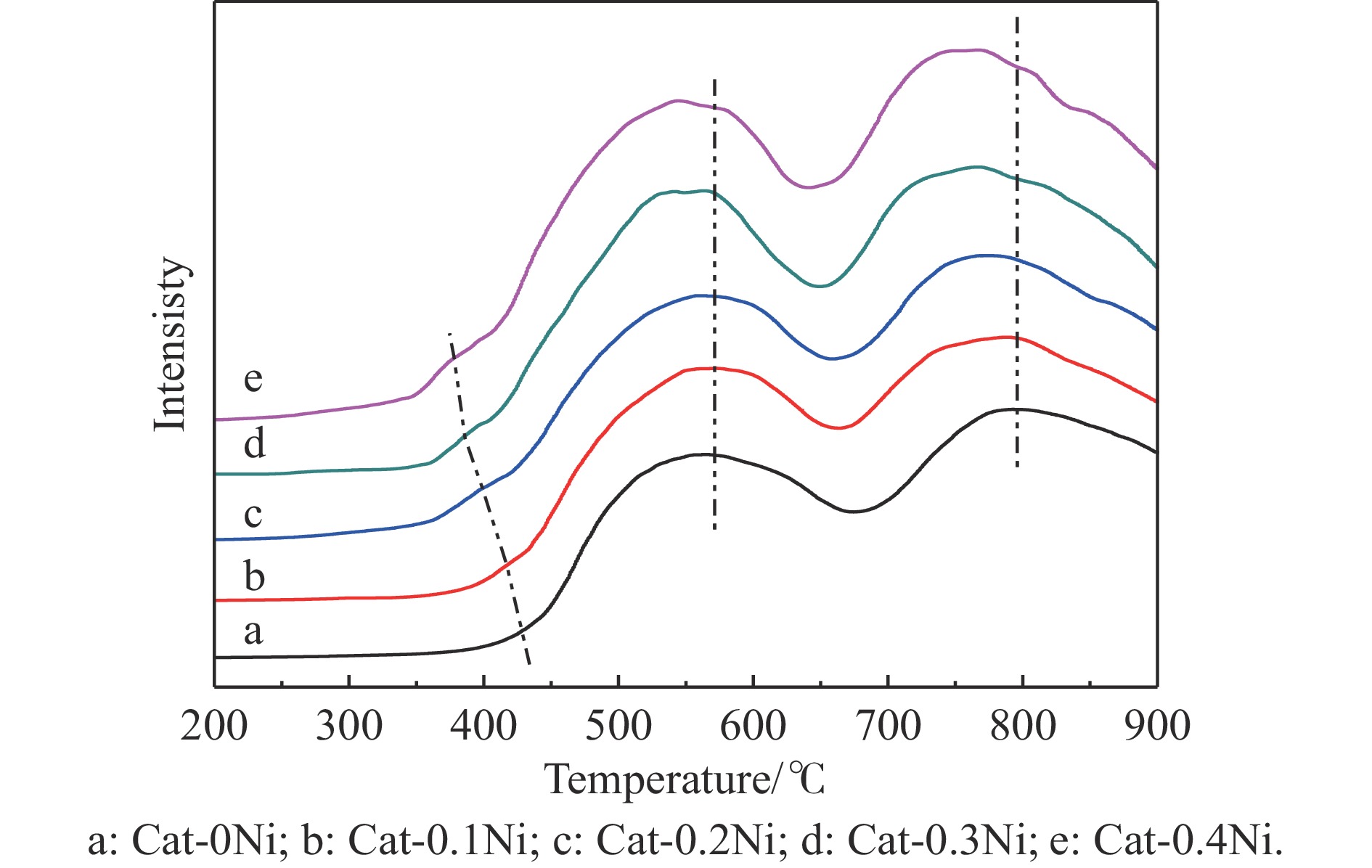

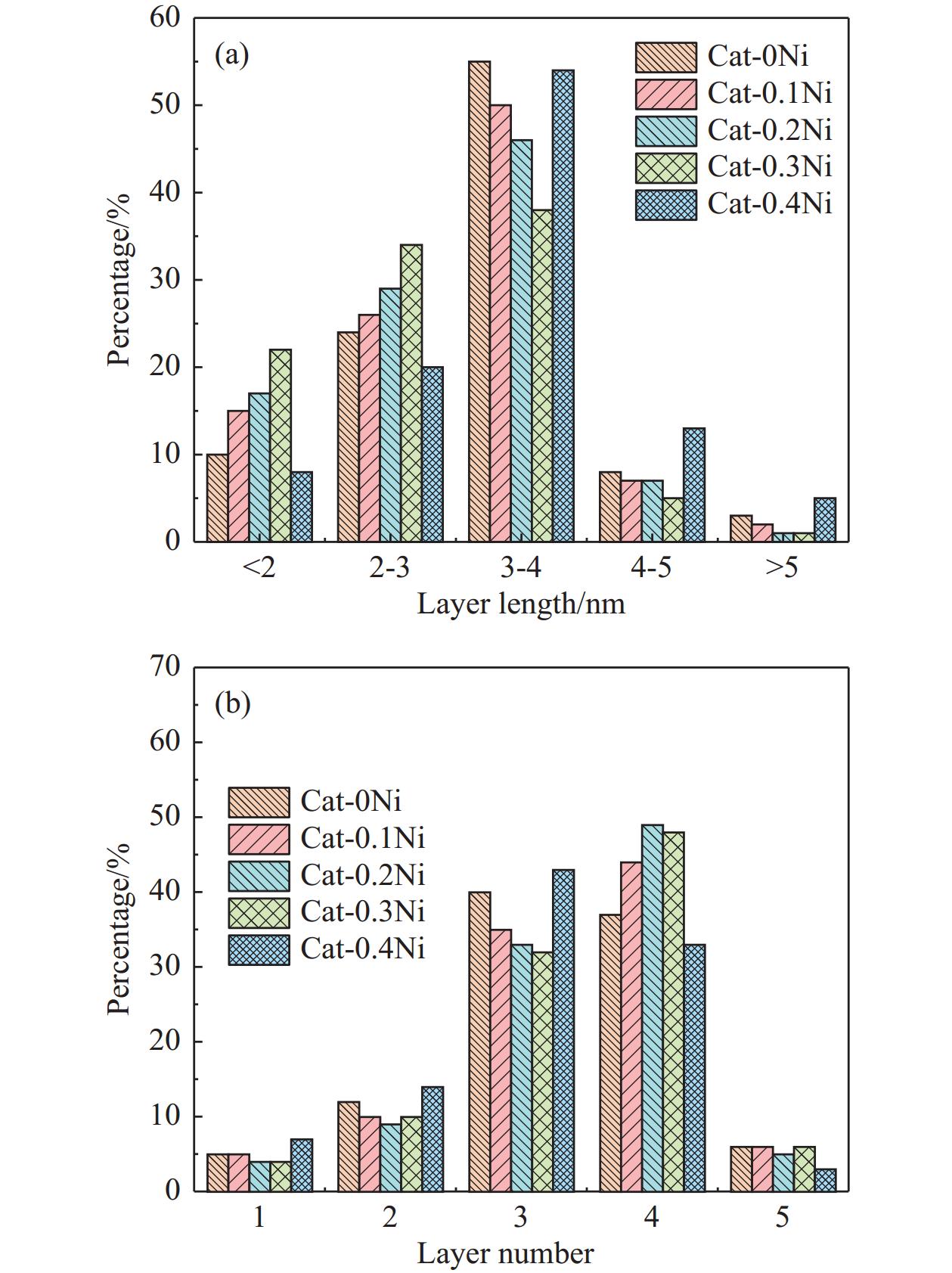
当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024015
摘要:
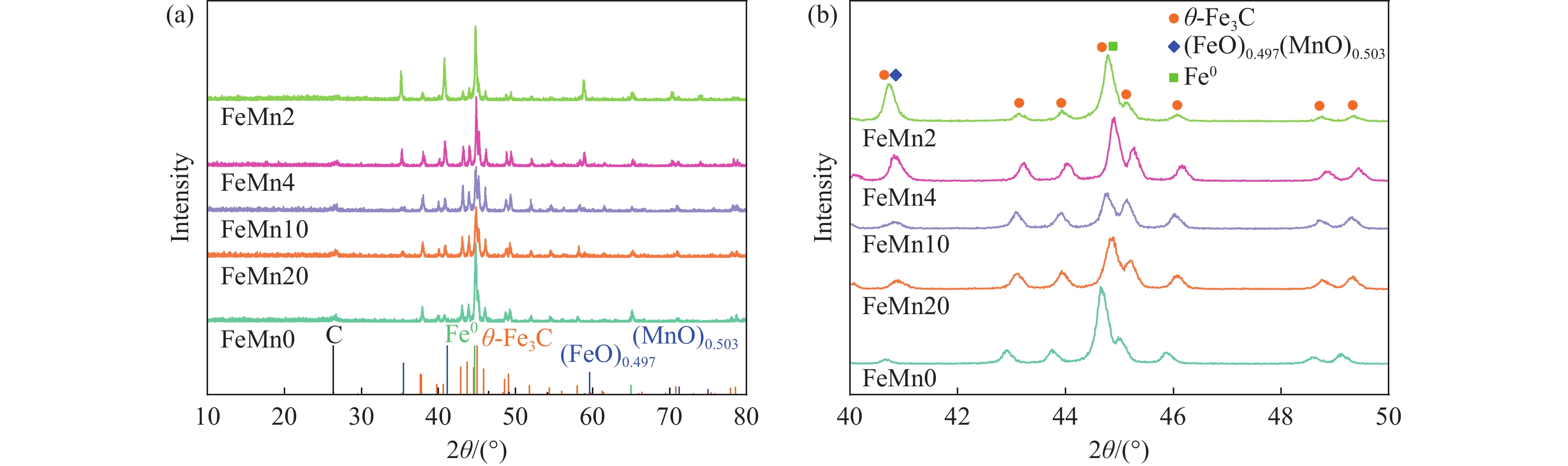
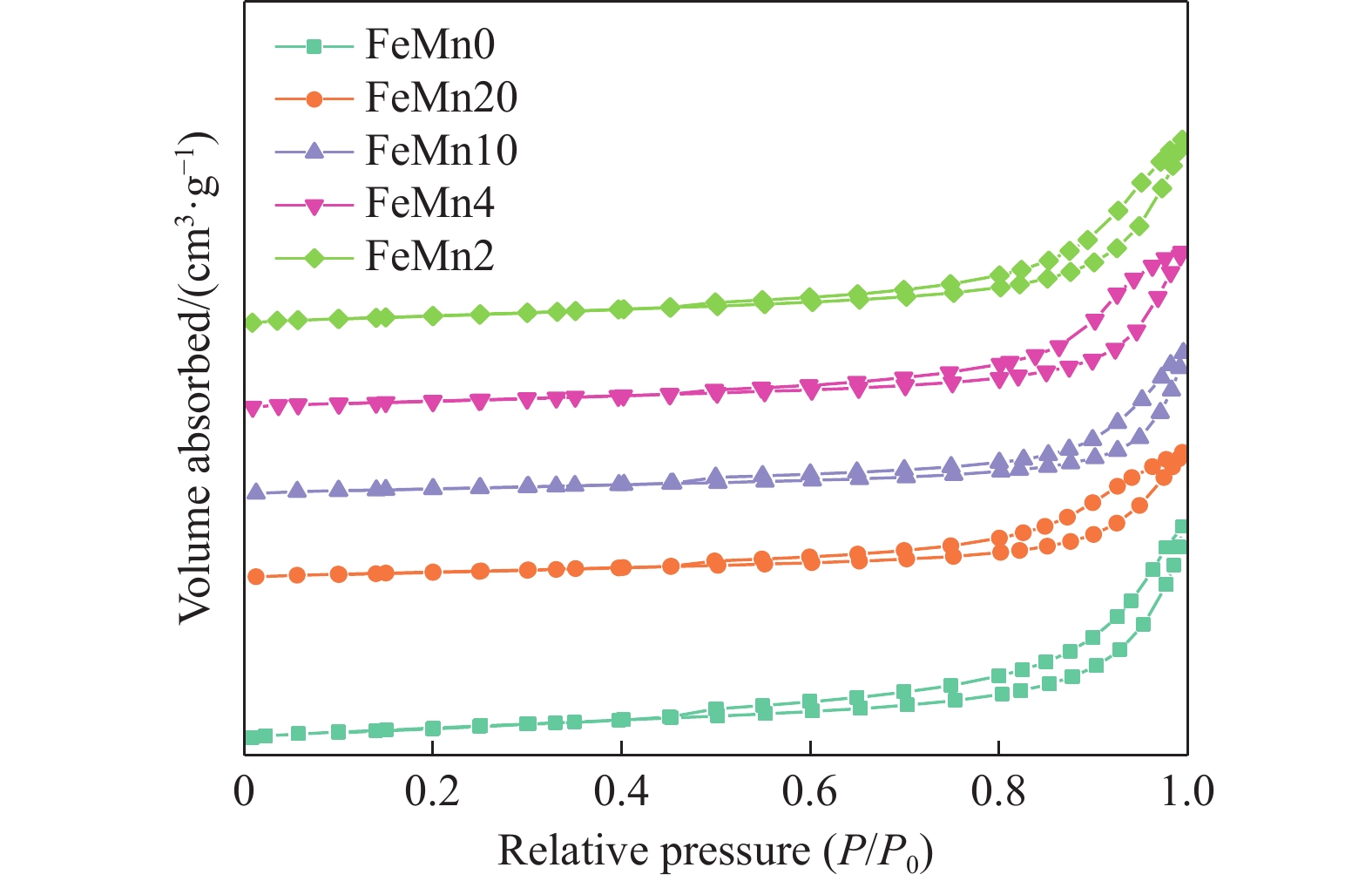
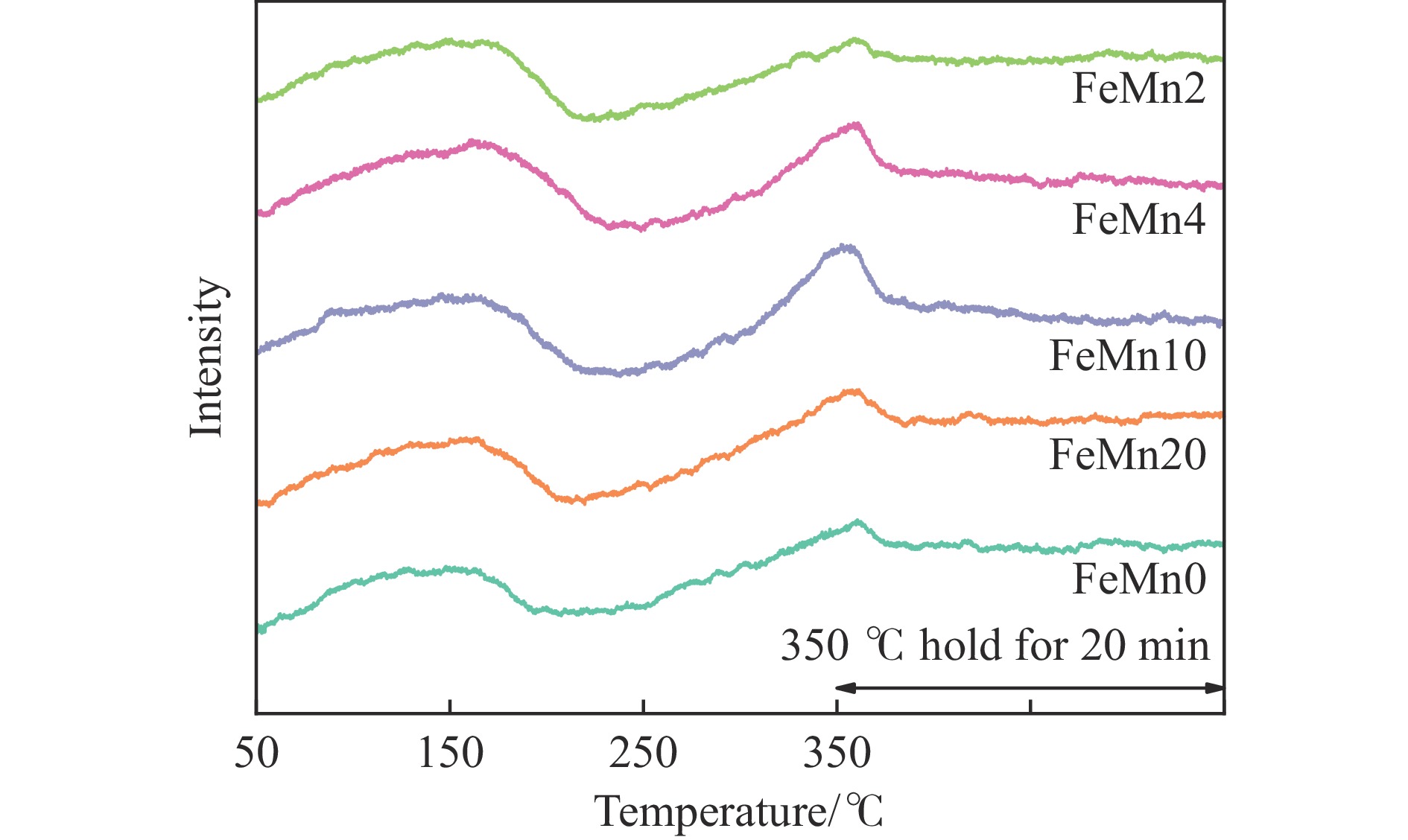
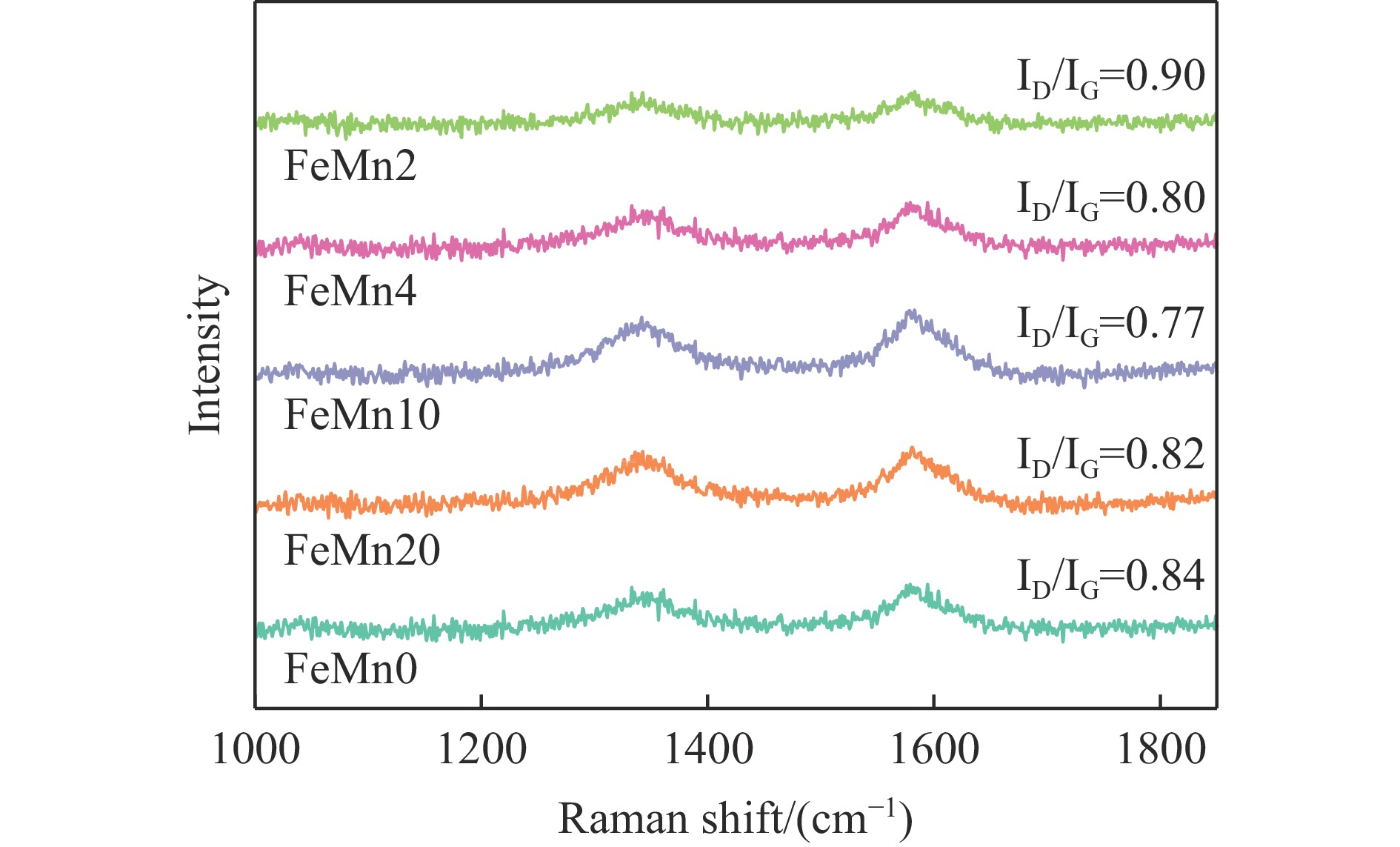
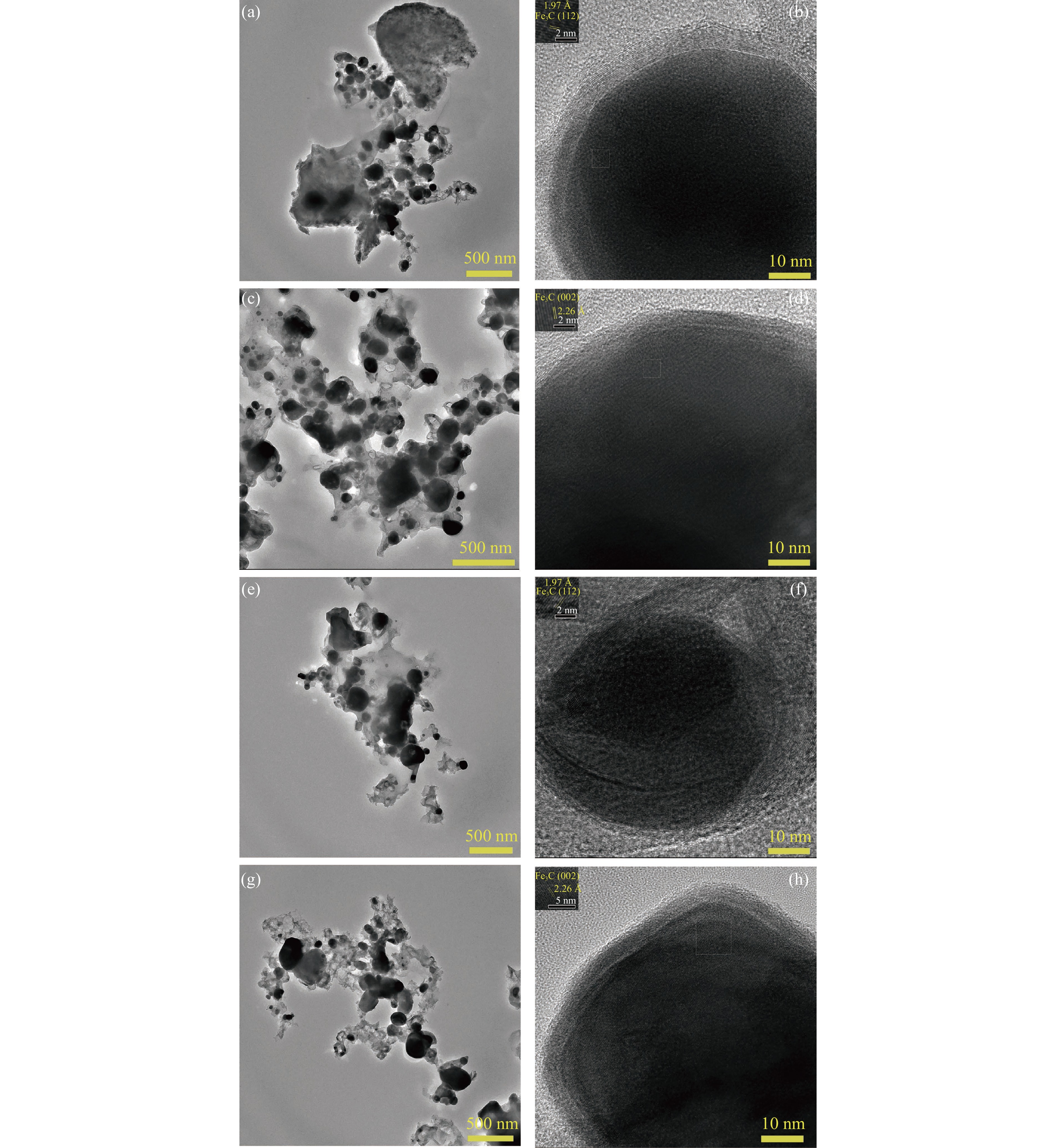
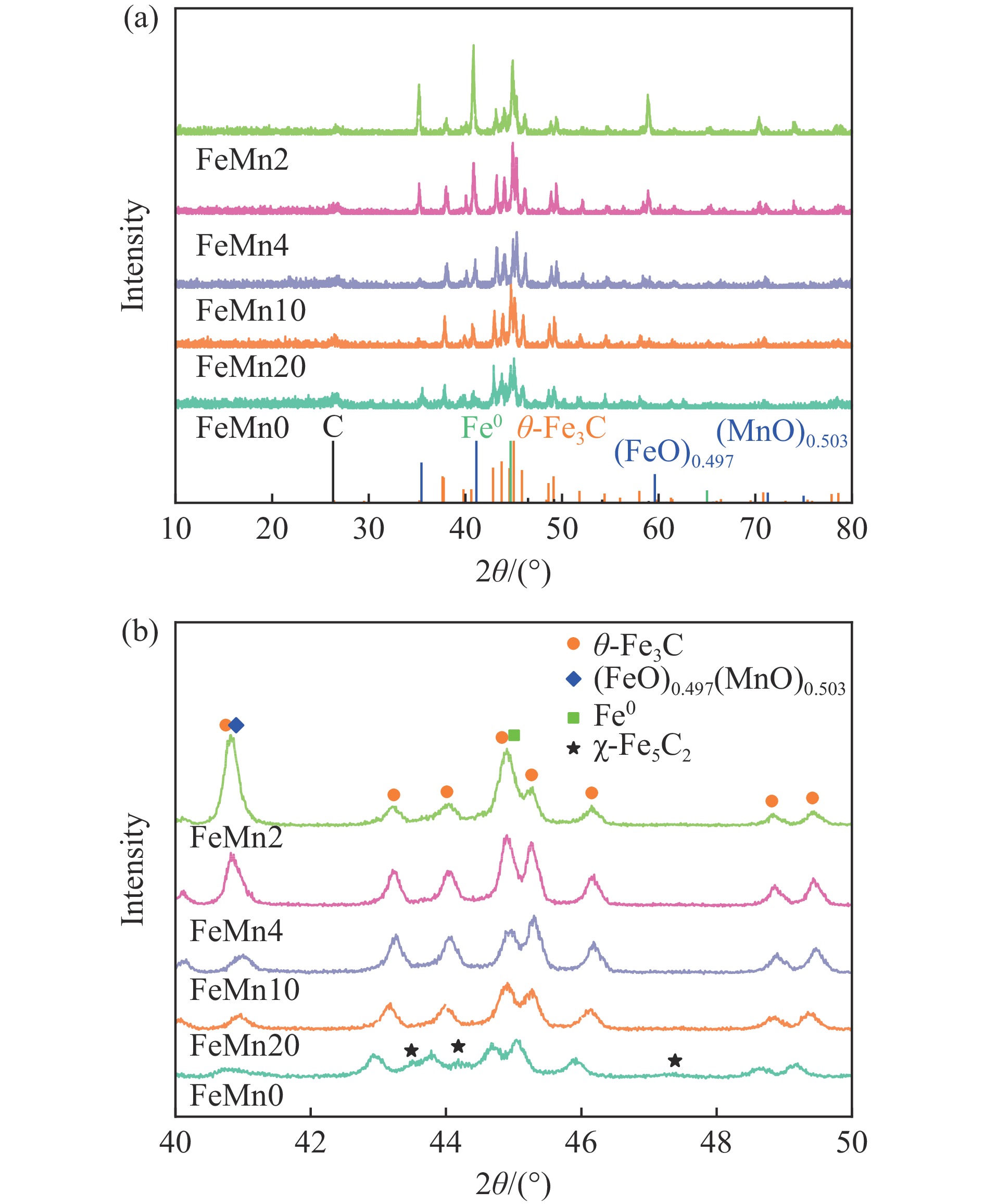
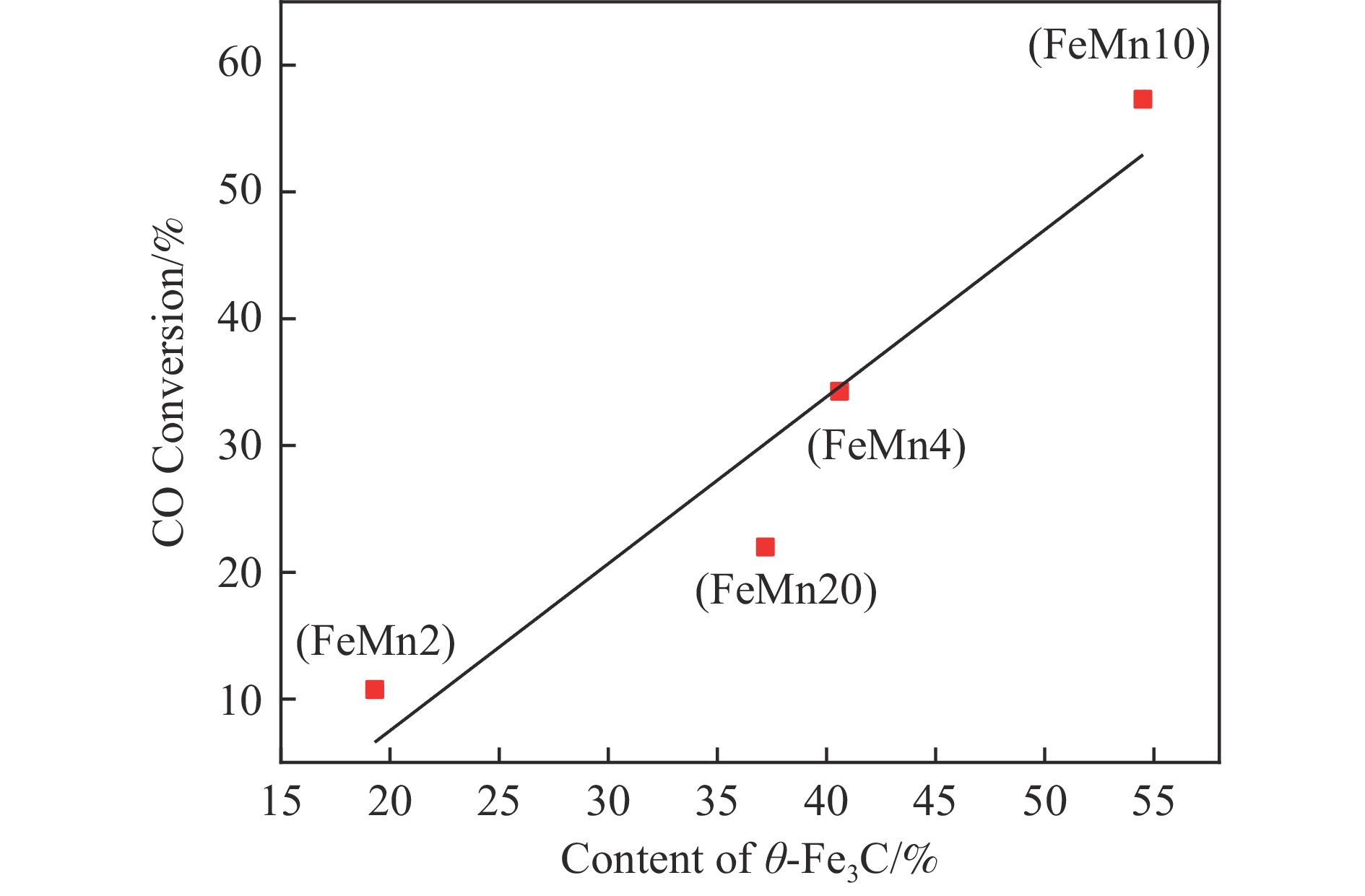
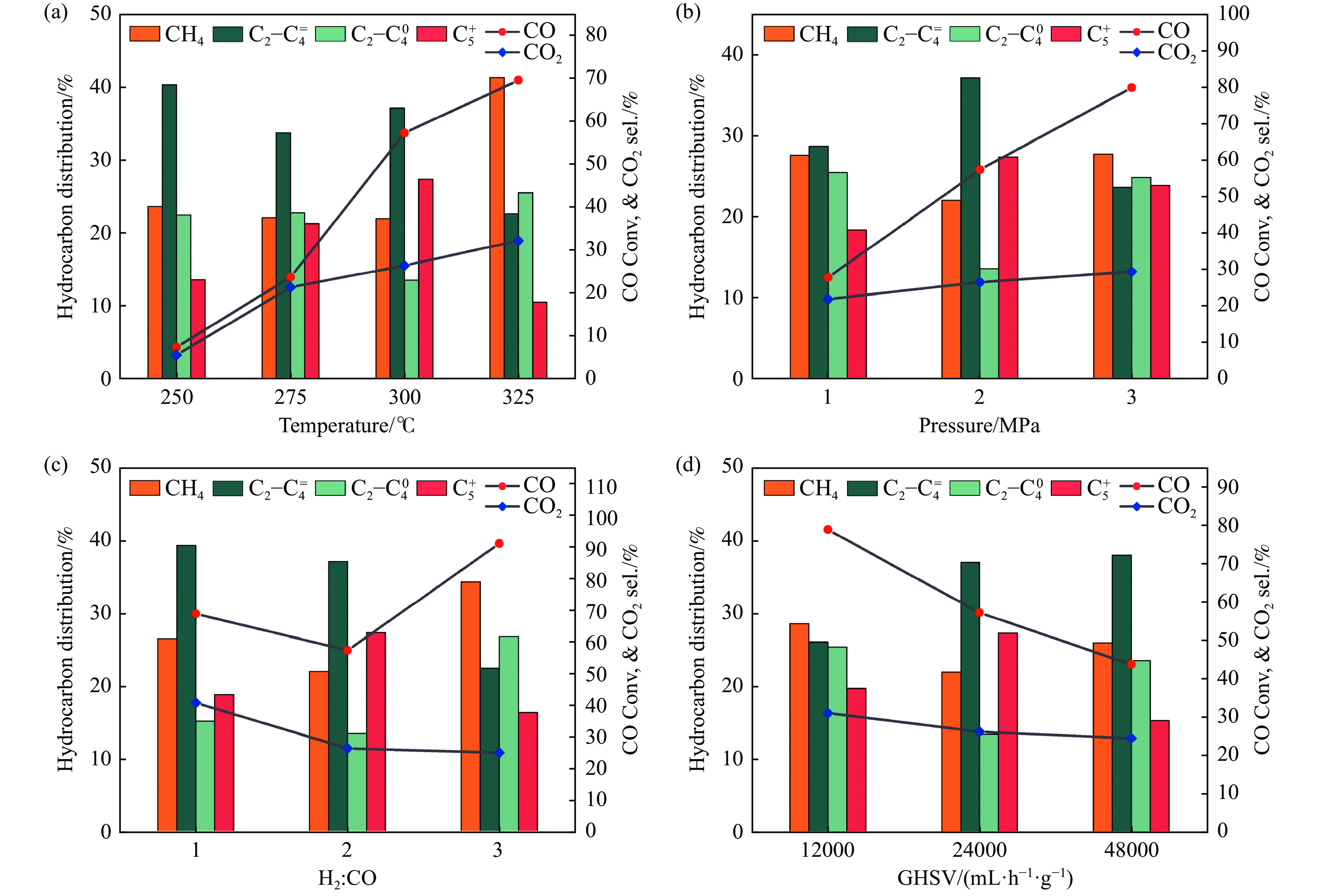
论文制备了系列含Fe、Mn的有机凝胶前驱体,在氩气氛围下通过高温热处理,凝胶中铁物种被有机物原位分解进行还原和碳化,制备出了θ-Fe3C含量不同的费托合成催化剂。采用XRD、N2吸附、Raman、CO-TPD、CO2-TPD、XPS和TEM等手段对催化剂的结构组成、表面性质以及活性物种的电子价态进行了系统的表征和分析。实验结果表明,热处理后获得的催化剂含石墨碳、θ-Fe3C、Fe0和(FeO)0.497(MnO)0.503物相,费托反应后催化剂的结构保持稳定,物相种类不发生变化。系统地考察了反应条件对催化性能的影响,FeMn10催化剂具有较优的催化性能,CO转化率为57.3%,低碳烯烃(C2-C4)选择性为37.1%,其中θ-Fe3C物相作为催化活性位点,催化剂的活性和低碳烯烃的选择性与θ-Fe3C的含量具有正相关性。
论文制备了系列含Fe、Mn的有机凝胶前驱体,在氩气氛围下通过高温热处理,凝胶中铁物种被有机物原位分解进行还原和碳化,制备出了θ-Fe3C含量不同的费托合成催化剂。采用XRD、N2吸附、Raman、CO-TPD、CO2-TPD、XPS和TEM等手段对催化剂的结构组成、表面性质以及活性物种的电子价态进行了系统的表征和分析。实验结果表明,热处理后获得的催化剂含石墨碳、θ-Fe3C、Fe0和(FeO)0.497(MnO)0.503物相,费托反应后催化剂的结构保持稳定,物相种类不发生变化。系统地考察了反应条件对催化性能的影响,FeMn10催化剂具有较优的催化性能,CO转化率为57.3%,低碳烯烃(C2-C4)选择性为37.1%,其中θ-Fe3C物相作为催化活性位点,催化剂的活性和低碳烯烃的选择性与θ-Fe3C的含量具有正相关性。



当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60446-9
摘要:
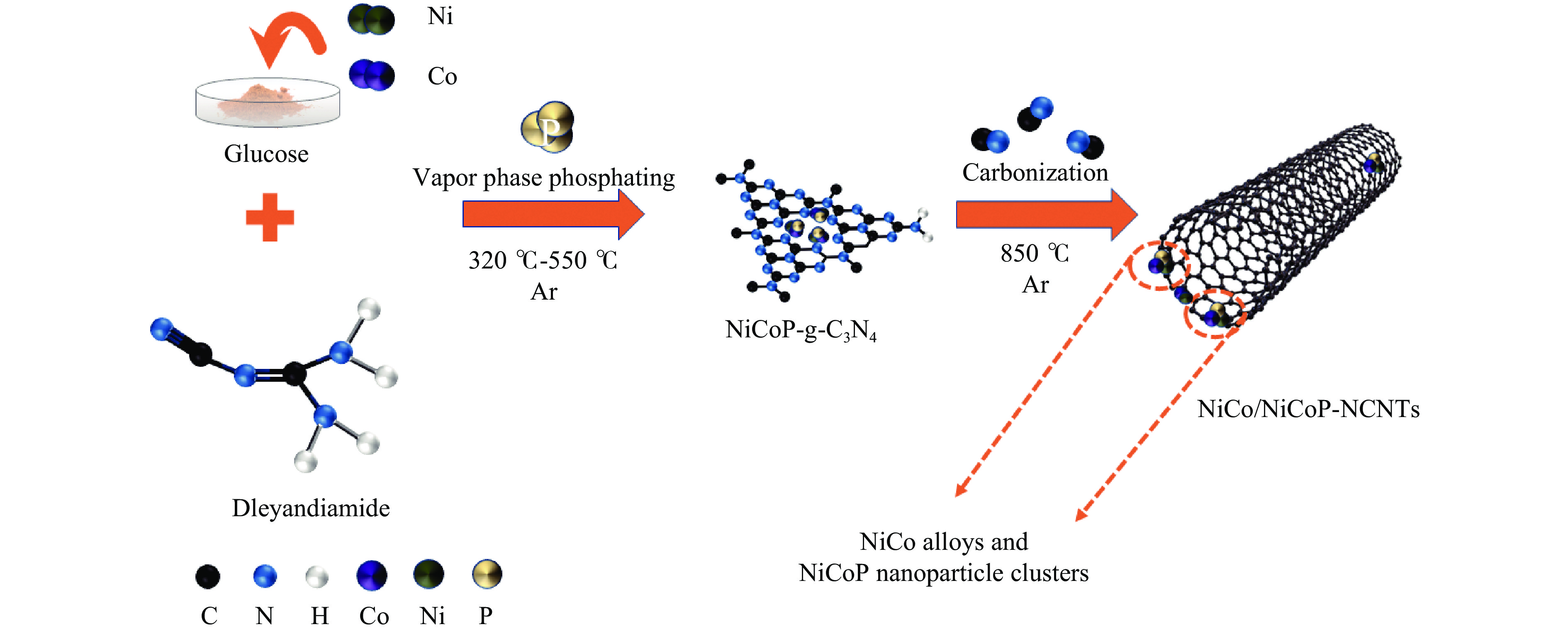
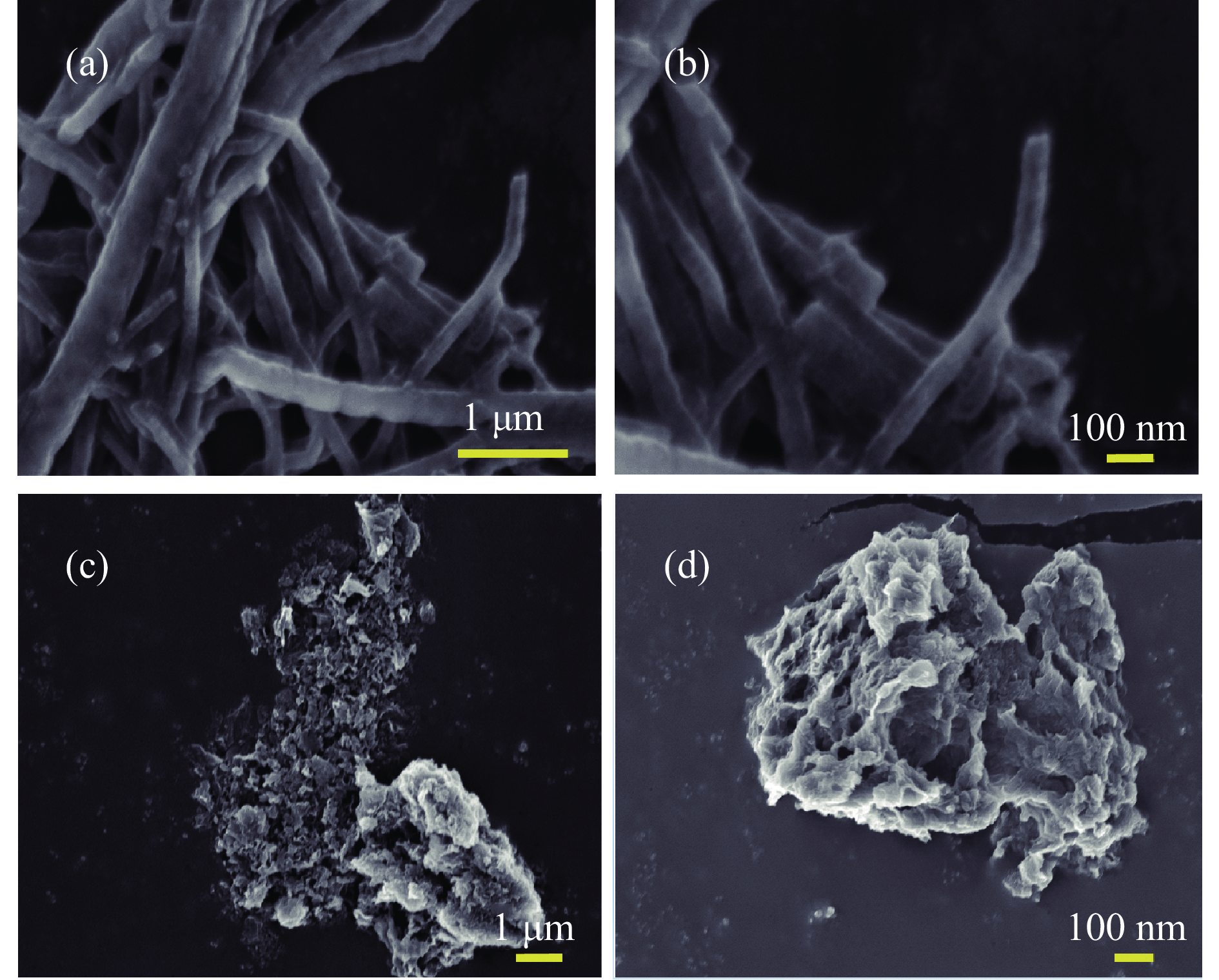
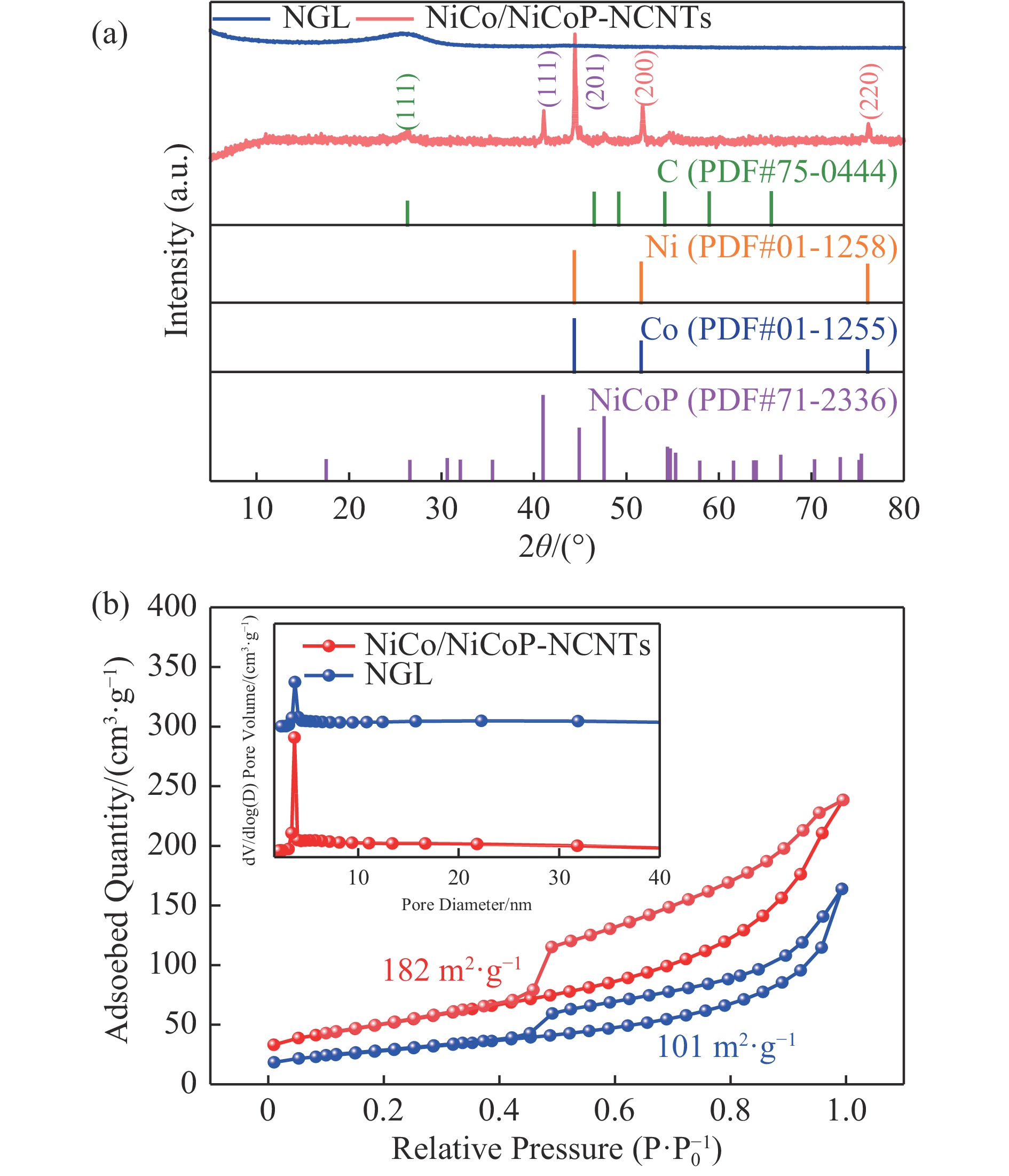
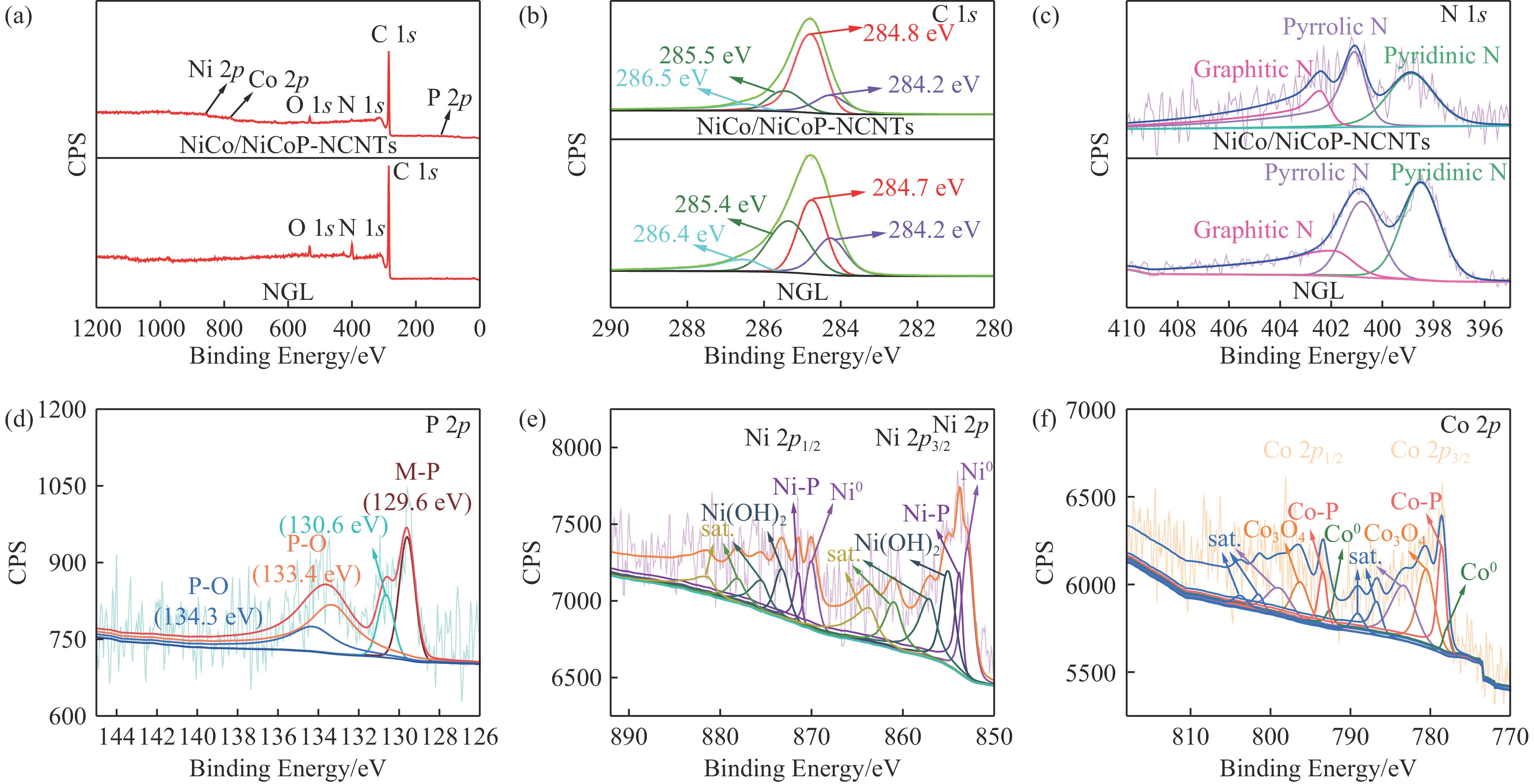
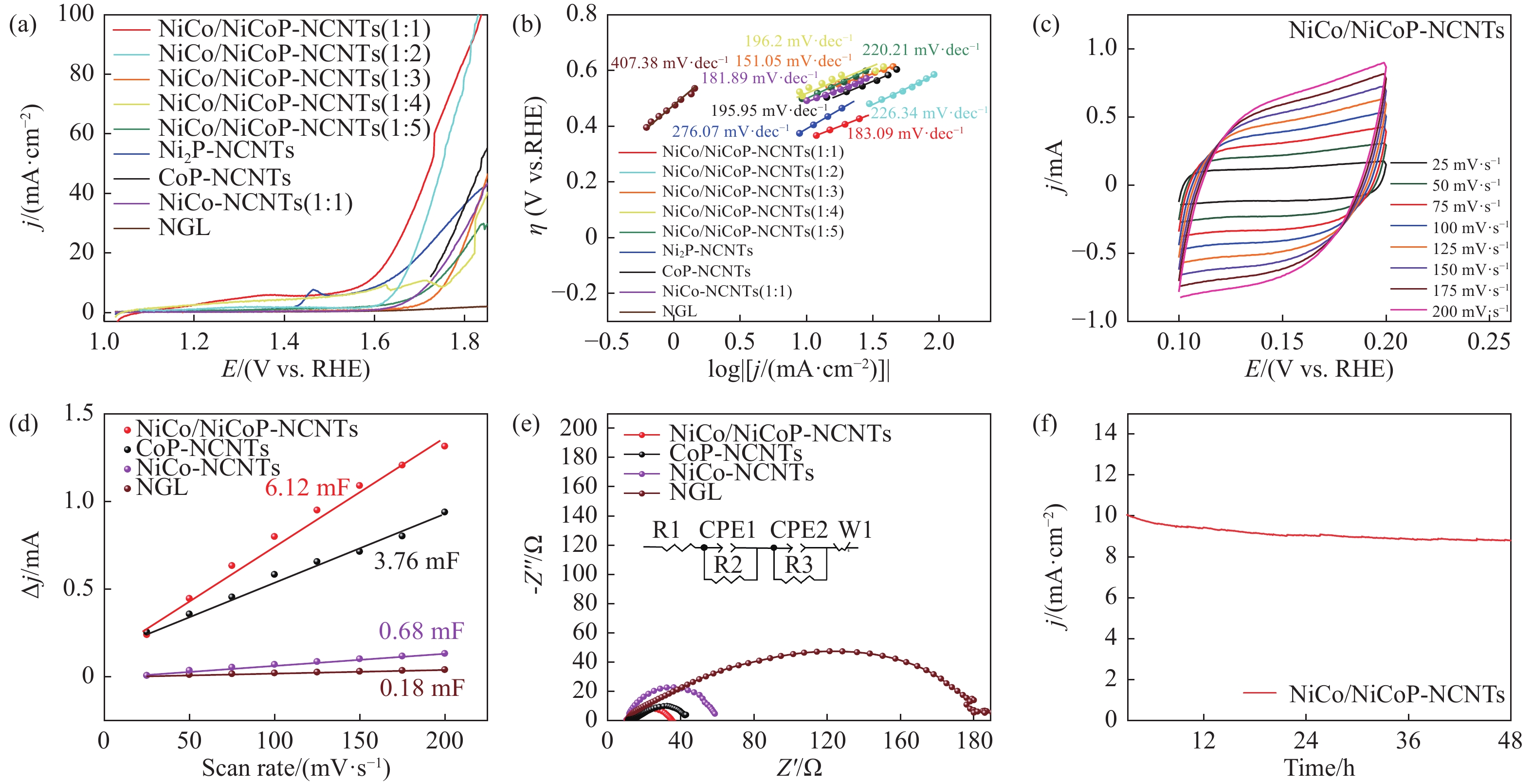
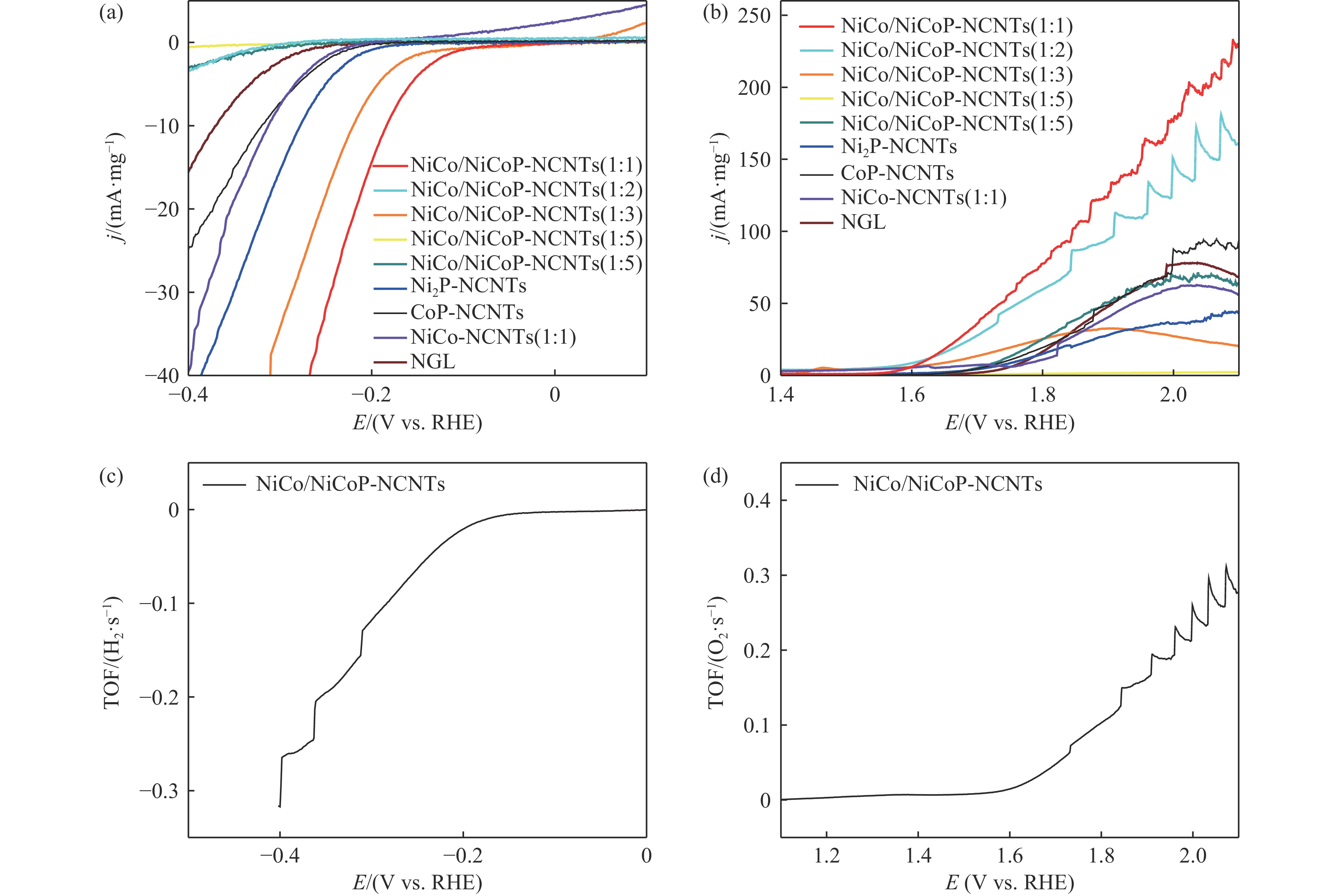
Ni, Co-induced highly distributed NiCoP nanoparticles embedded nitrogen-doped carbon nanotubes (NCNTs) (NiCo/NiCoP-NCNTs) were directly synthesized by a one-step phosphorization and carbonization process. As a bifunctional electrocatalyst for water splitting, NiCo/NiCoP NCNTs show impressive catalytic performance with an overpotential of only 206 mV for the hydrogen evolution reaction and 360 mV for the oxygen evolution reaction in 0.5 M H2SO4 and 1 M KOH solutions, respectively. In addition, NiCo/NiCoP NCNTs maintain a stable cell voltage of 1.68 V at 10 mA cm-2 with only a 10% decrease in current density over 48 hours, showing remarkable stability. The improved catalytic activity can be attributed to the integration of NiCoP nanoparticles and the synergies between NCNTs and NiCo alloy. Additionally, the improved electrocatalytic performance can be attributed to the increased electrochemically active surface area and the reduced electron transfer resistance of the NiCo/NiCoP-NCNTs. Overall, the NiCo/NiCoP-NCNTs demonstrated significant performance for advanced water electrolysis applications.
Ni, Co-induced highly distributed NiCoP nanoparticles embedded nitrogen-doped carbon nanotubes (NCNTs) (NiCo/NiCoP-NCNTs) were directly synthesized by a one-step phosphorization and carbonization process. As a bifunctional electrocatalyst for water splitting, NiCo/NiCoP NCNTs show impressive catalytic performance with an overpotential of only 206 mV for the hydrogen evolution reaction and 360 mV for the oxygen evolution reaction in 0.5 M H2SO4 and 1 M KOH solutions, respectively. In addition, NiCo/NiCoP NCNTs maintain a stable cell voltage of 1.68 V at 10 mA cm-2 with only a 10% decrease in current density over 48 hours, showing remarkable stability. The improved catalytic activity can be attributed to the integration of NiCoP nanoparticles and the synergies between NCNTs and NiCo alloy. Additionally, the improved electrocatalytic performance can be attributed to the increased electrochemically active surface area and the reduced electron transfer resistance of the NiCo/NiCoP-NCNTs. Overall, the NiCo/NiCoP-NCNTs demonstrated significant performance for advanced water electrolysis applications.


当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023089
摘要:
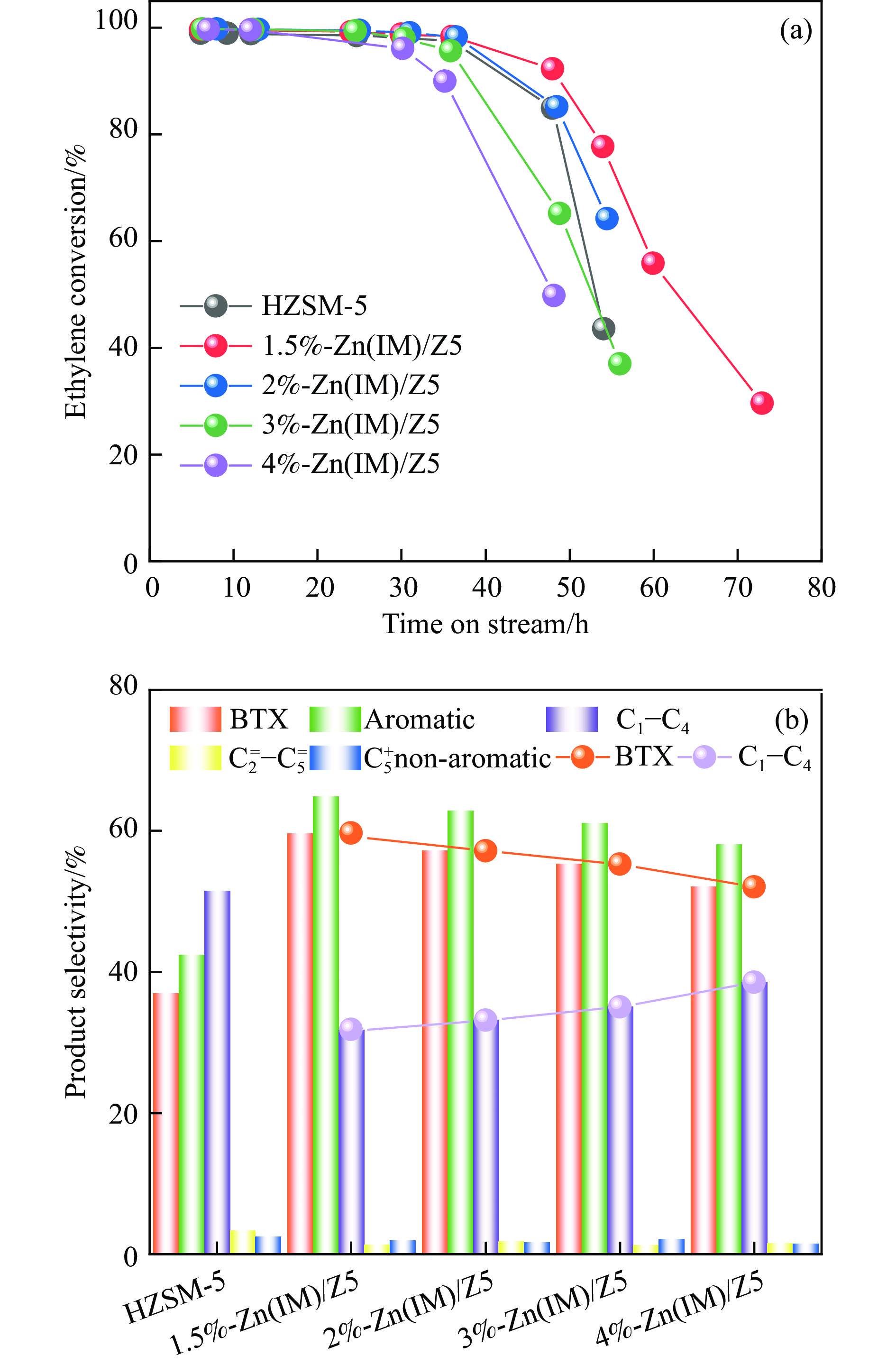
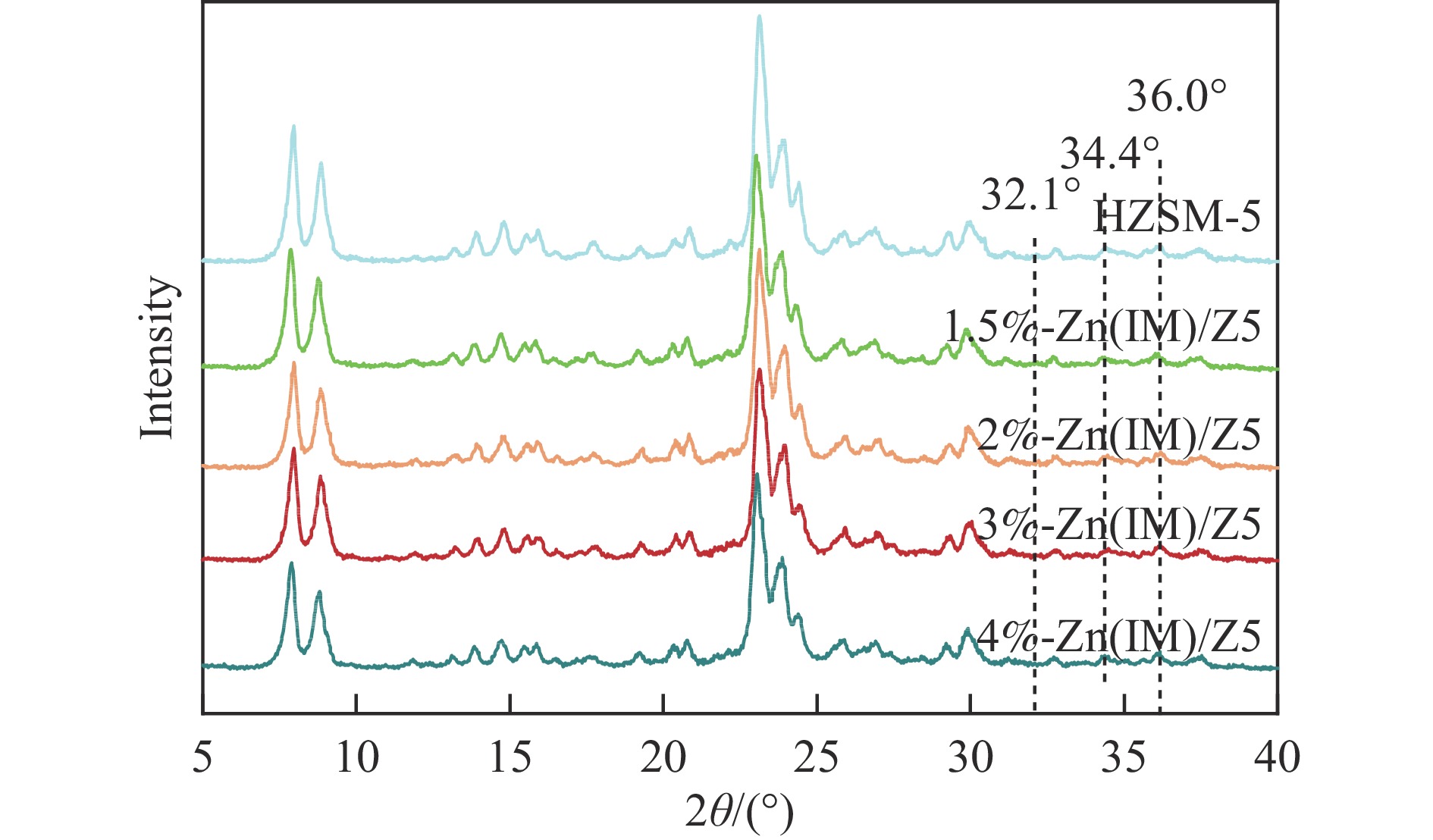
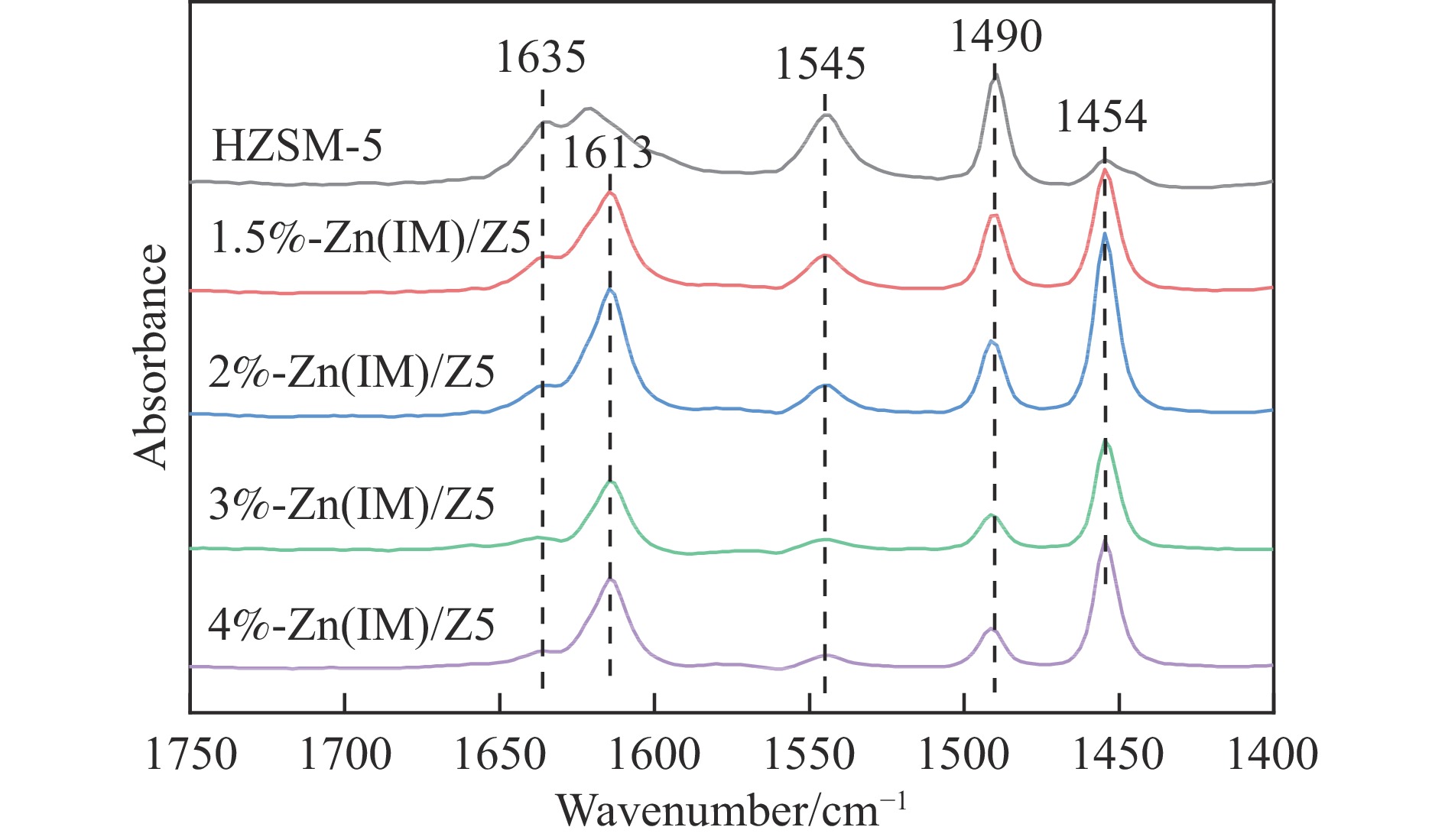
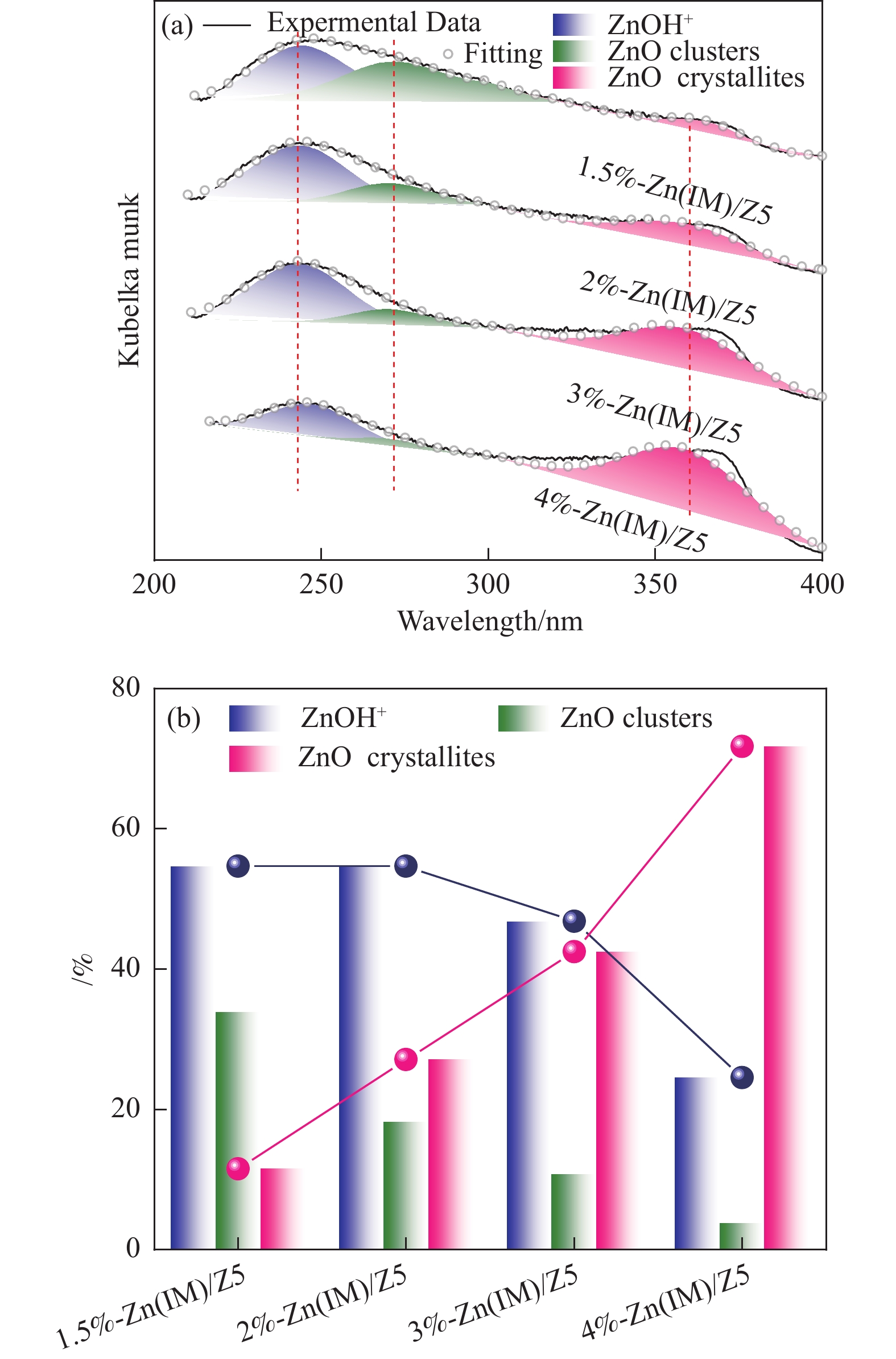
HZSM-5(Si/Al=30)为载体,用等体积浸渍法合成了系列不同Zn负载量的双功能Zn/ZSM-5催化剂,考察了Zn负载量在乙烯芳构化过程中的催化性能。用X射线粉末衍射(XRD)、N2吸附-脱附和吡啶吸附红外光谱(Py-FTIR)方法考察了催化剂的结构和酸性,用电感耦合等离子发射光谱(ICP)、紫外可见光谱(UV-vis DRS)、X射线吸收精细结构(XAFS)技术解析了Zn物种的结构及其流失行为。结果表明,Zn含量对其在HZSM-5上的存在状态及催化乙烯芳构化反应性能均有明显的影响,具有较多活性六配位ZnOH+物种的1.5%-Zn(IM)/Z5催化剂表现出较高的芳烃选择性和催化剂稳定性,且表现出较低的Zn流失速率。
HZSM-5(Si/Al=30)为载体,用等体积浸渍法合成了系列不同Zn负载量的双功能Zn/ZSM-5催化剂,考察了Zn负载量在乙烯芳构化过程中的催化性能。用X射线粉末衍射(XRD)、N2吸附-脱附和吡啶吸附红外光谱(Py-FTIR)方法考察了催化剂的结构和酸性,用电感耦合等离子发射光谱(ICP)、紫外可见光谱(UV-vis DRS)、X射线吸收精细结构(XAFS)技术解析了Zn物种的结构及其流失行为。结果表明,Zn含量对其在HZSM-5上的存在状态及催化乙烯芳构化反应性能均有明显的影响,具有较多活性六配位ZnOH+物种的1.5%-Zn(IM)/Z5催化剂表现出较高的芳烃选择性和催化剂稳定性,且表现出较低的Zn流失速率。

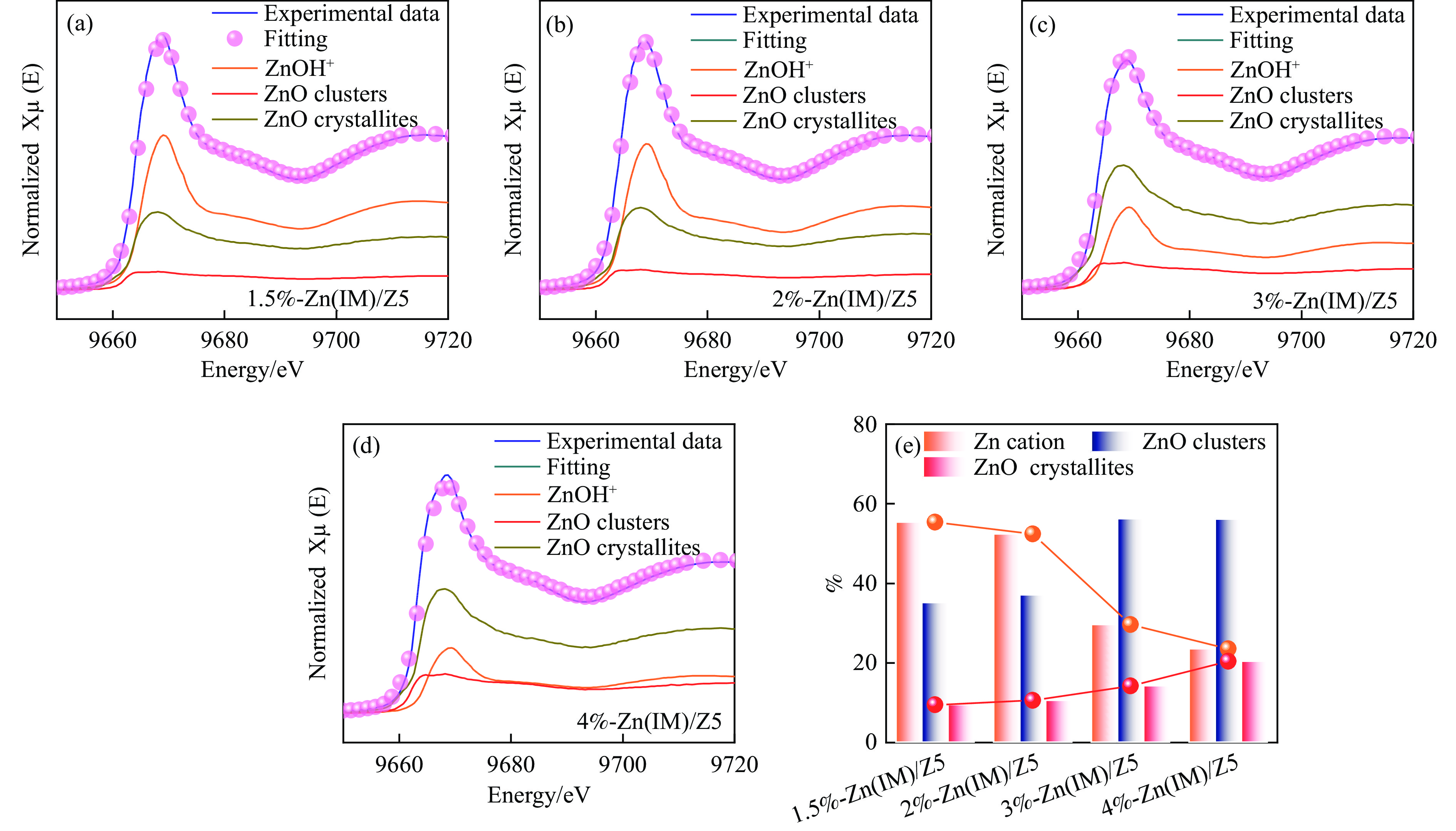

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023083
摘要:
火焰喷雾热解法(FSP)是一种简单、快速、可规模化制备纳米催化剂的技术。通过火焰喷雾热解法合成CeO2和Pt-CeO2载体、Pd-Pt-CeO2催化剂,采用浸渍法在CeO2和Pt-CeO2载体分别沉积Pd-Pt和Pd而制得Pd-Pt双金属催化剂,并考察其甲烷催化燃烧性能。利用ICP、XRD、TEM、BET、H2-TPR、XPS和Raman对催化剂的物化性质进行分析。TEM结果表明,Pd-Pt/CeO2催化剂中Pd和Pt物种高分散于CeO2载体。相比于一步法(one step)制得的Pd-Pt-CeO2(OS-FSP)催化剂,共浸渍法制得Pd-Pt/CeO2(0.25)-WI的催化活性更高,其t50降低了60 ℃,且稳定运行60 h而没有明显失活。这归因于Pd-Pt/CeO2(0.25)-WI催化剂表面上Pd0/Pd2+和Ce3+/Ce4+物质的量比更高、晶格氧更多,进而导致其具有良好的甲烷催化燃烧性能。
火焰喷雾热解法(FSP)是一种简单、快速、可规模化制备纳米催化剂的技术。通过火焰喷雾热解法合成CeO2和Pt-CeO2载体、Pd-Pt-CeO2催化剂,采用浸渍法在CeO2和Pt-CeO2载体分别沉积Pd-Pt和Pd而制得Pd-Pt双金属催化剂,并考察其甲烷催化燃烧性能。利用ICP、XRD、TEM、BET、H2-TPR、XPS和Raman对催化剂的物化性质进行分析。TEM结果表明,Pd-Pt/CeO2催化剂中Pd和Pt物种高分散于CeO2载体。相比于一步法(one step)制得的Pd-Pt-CeO2(OS-FSP)催化剂,共浸渍法制得Pd-Pt/CeO2(0.25)-WI的催化活性更高,其t50降低了60 ℃,且稳定运行60 h而没有明显失活。这归因于Pd-Pt/CeO2(0.25)-WI催化剂表面上Pd0/Pd2+和Ce3+/Ce4+物质的量比更高、晶格氧更多,进而导致其具有良好的甲烷催化燃烧性能。









当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023079
摘要:
采用废弃的鸡蛋壳作载体,沉积沉淀法制备了一系列不同Co3O4含量Co3O4/鸡蛋壳催化剂,并在连续流动微反装置上考察了N2O分解性能。结果表明,当Co3O4质量分数为20%时,催化剂表现出优异的N2O分解性能。在空速10000 h−1和N2O含量0.1%的条件下,400 ℃可实现N2O完全转化;其比活性约为Co3O4催化剂的4.3倍(反应温度为440 ℃);同时,该催化剂对原料气中3% O2、3.3% H2O和/或2.0×10−4 NO表现出较强的耐受性和较高的稳定性。分析催化剂的多种表征结果发现,CaCO3作为鸡蛋壳的主要成分,与活性组分Co3O4紧密结合,两者的强相互作用导致20%Co3O4/鸡蛋壳催化剂中产生更多的氧空位和Co3+;Co3O4氧化还原性能得到提高,Co−O键被有效削弱;此外,该强相互作用可提高20%Co3O4/鸡蛋壳催化剂表面碱性位点的强度,增大碱性位点数量,更易于转移电子而促进N2O分解。
采用废弃的鸡蛋壳作载体,沉积沉淀法制备了一系列不同Co3O4含量Co3O4/鸡蛋壳催化剂,并在连续流动微反装置上考察了N2O分解性能。结果表明,当Co3O4质量分数为20%时,催化剂表现出优异的N2O分解性能。在空速10000 h−1和N2O含量0.1%的条件下,400 ℃可实现N2O完全转化;其比活性约为Co3O4催化剂的4.3倍(反应温度为440 ℃);同时,该催化剂对原料气中3% O2、3.3% H2O和/或2.0×10−4 NO表现出较强的耐受性和较高的稳定性。分析催化剂的多种表征结果发现,CaCO3作为鸡蛋壳的主要成分,与活性组分Co3O4紧密结合,两者的强相互作用导致20%Co3O4/鸡蛋壳催化剂中产生更多的氧空位和Co3+;Co3O4氧化还原性能得到提高,Co−O键被有效削弱;此外,该强相互作用可提高20%Co3O4/鸡蛋壳催化剂表面碱性位点的强度,增大碱性位点数量,更易于转移电子而促进N2O分解。












当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60400-1
摘要:
Cu2O具有禁带窄、环境友好和储量丰富等优点,是一种理想的可见光催化剂,然而其光生载流子复合率高和稳定性差等问题限制了Cu2O在光催化领域的实际应用。为此,本文采用光诱导原位技术,以甲醇为碳源、硫酸铜为铜源,一步成功制备了超薄碳壳层包覆的Cu2O复合纳米材料(Cu2O@C)。结果显示,与常规碳包覆方法相比,光诱导原位技术避免了苛刻的反应条件及繁琐的合成步骤对Cu2O半导体结构的破坏,有效保留了Cu2O本征电子结构,使其具有优异的光催化活性及稳定性。同时,Cu2O@C的核壳结构不仅可以钝化半导体表面缺陷和促进光生载流子的分离,而且碳壳层的包覆还可以避免Cu2O纳米颗粒与溶液的直接接触,有效抑制高活性反应中间体对催化剂结构的破坏。与单独的Cu2O纳米颗粒相比,Cu2O@C复合纳米材料在可见光下的光解水产氢活性和稳定性得到显著提高,产氢速率可达1.28 mmol/(g·h),且在连续五次循环稳定性测试中,氢气生成速率无明显变化。
Cu2O具有禁带窄、环境友好和储量丰富等优点,是一种理想的可见光催化剂,然而其光生载流子复合率高和稳定性差等问题限制了Cu2O在光催化领域的实际应用。为此,本文采用光诱导原位技术,以甲醇为碳源、硫酸铜为铜源,一步成功制备了超薄碳壳层包覆的Cu2O复合纳米材料(Cu2O@C)。结果显示,与常规碳包覆方法相比,光诱导原位技术避免了苛刻的反应条件及繁琐的合成步骤对Cu2O半导体结构的破坏,有效保留了Cu2O本征电子结构,使其具有优异的光催化活性及稳定性。同时,Cu2O@C的核壳结构不仅可以钝化半导体表面缺陷和促进光生载流子的分离,而且碳壳层的包覆还可以避免Cu2O纳米颗粒与溶液的直接接触,有效抑制高活性反应中间体对催化剂结构的破坏。与单独的Cu2O纳米颗粒相比,Cu2O@C复合纳米材料在可见光下的光解水产氢活性和稳定性得到显著提高,产氢速率可达1.28 mmol/(g·h),且在连续五次循环稳定性测试中,氢气生成速率无明显变化。








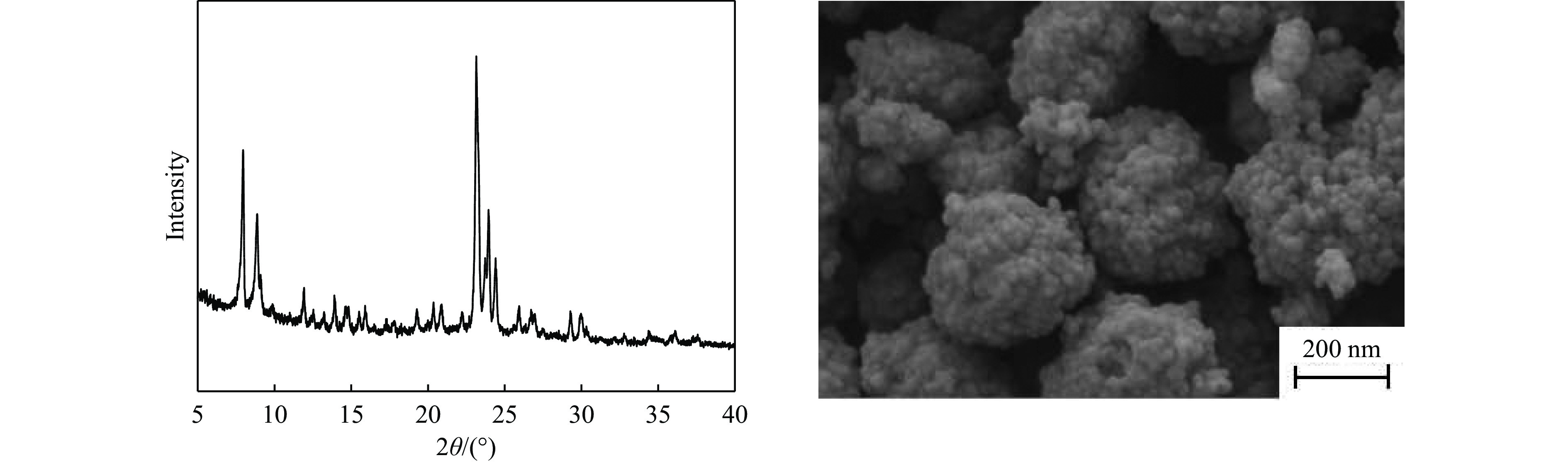
当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60443-3
摘要:
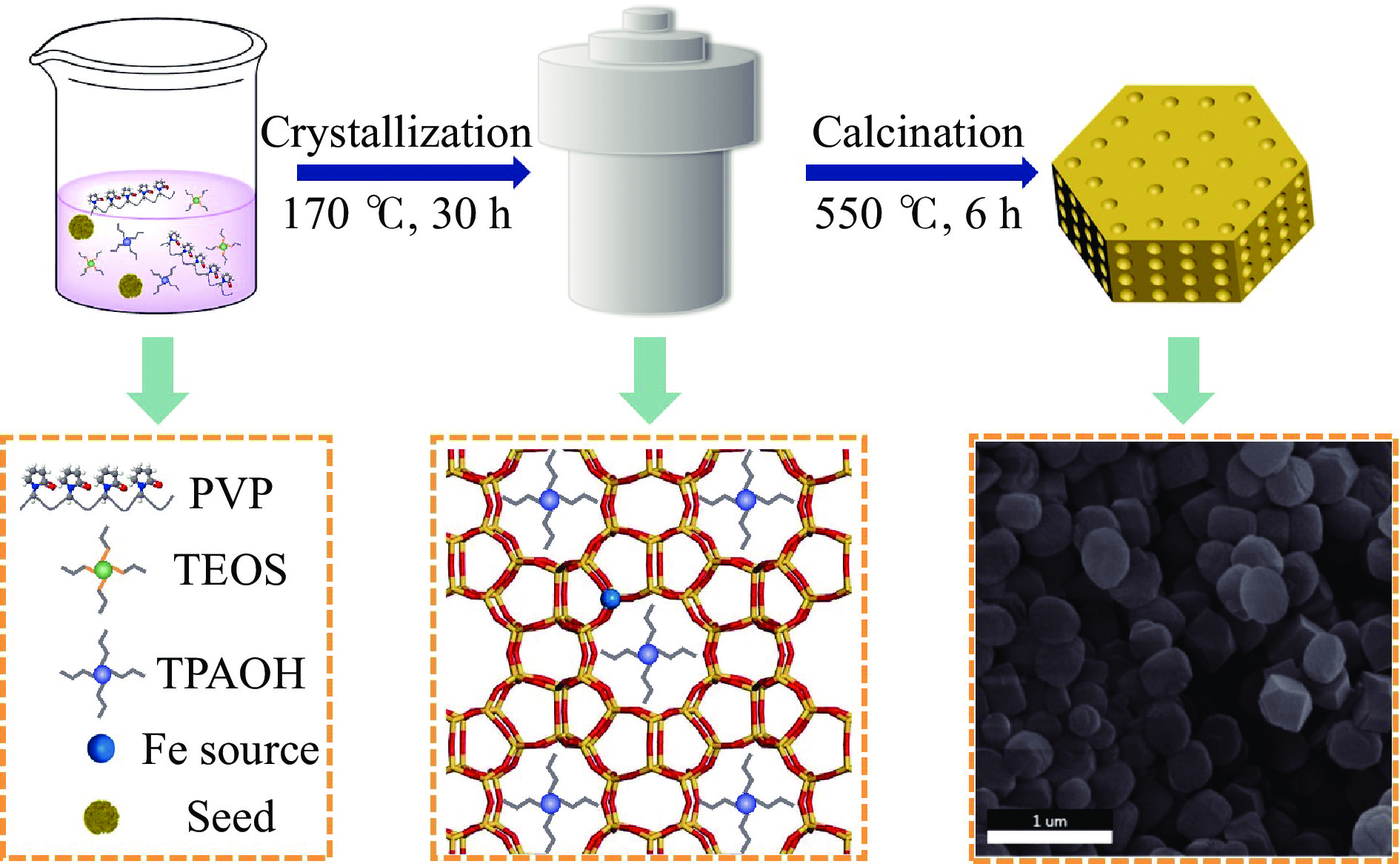
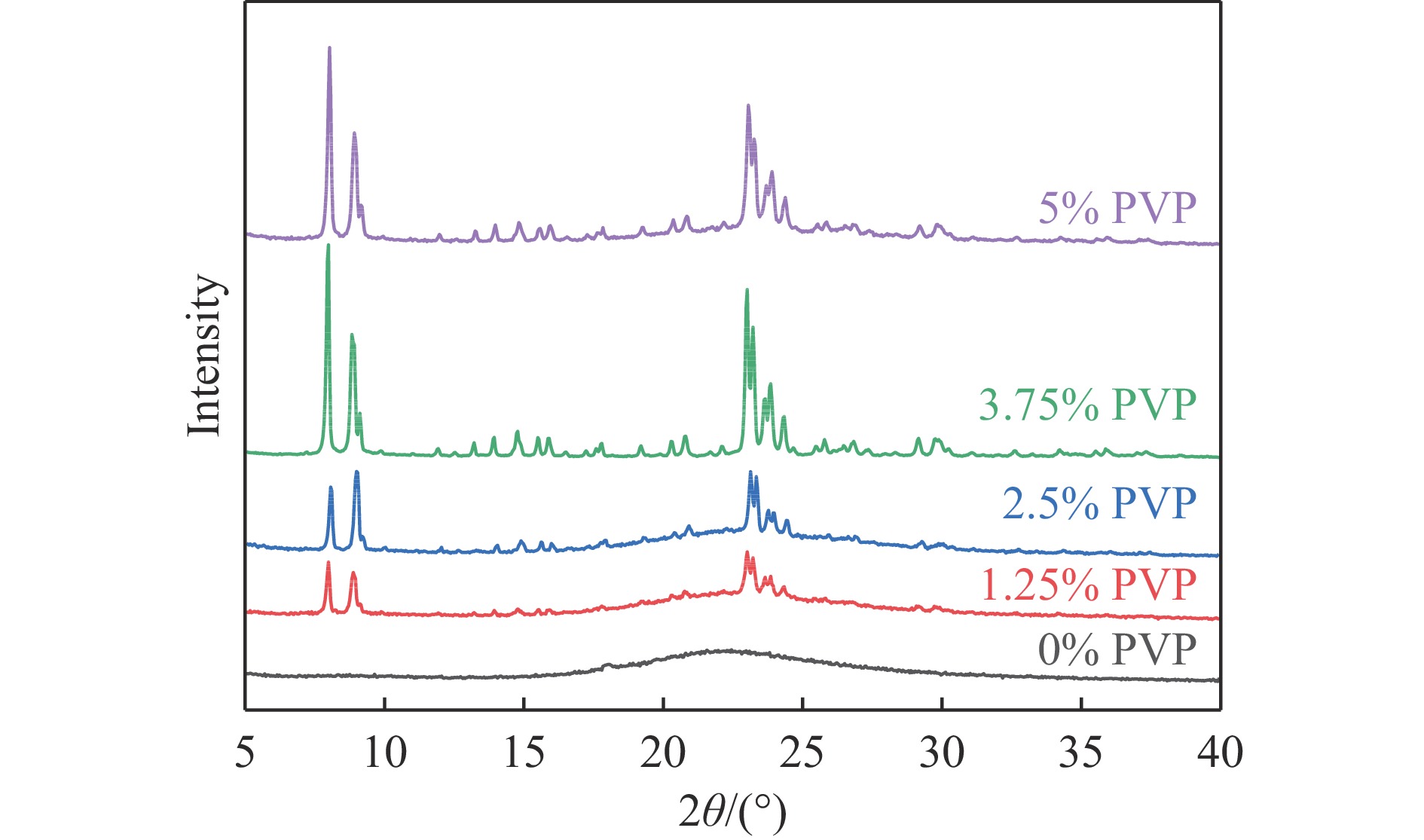
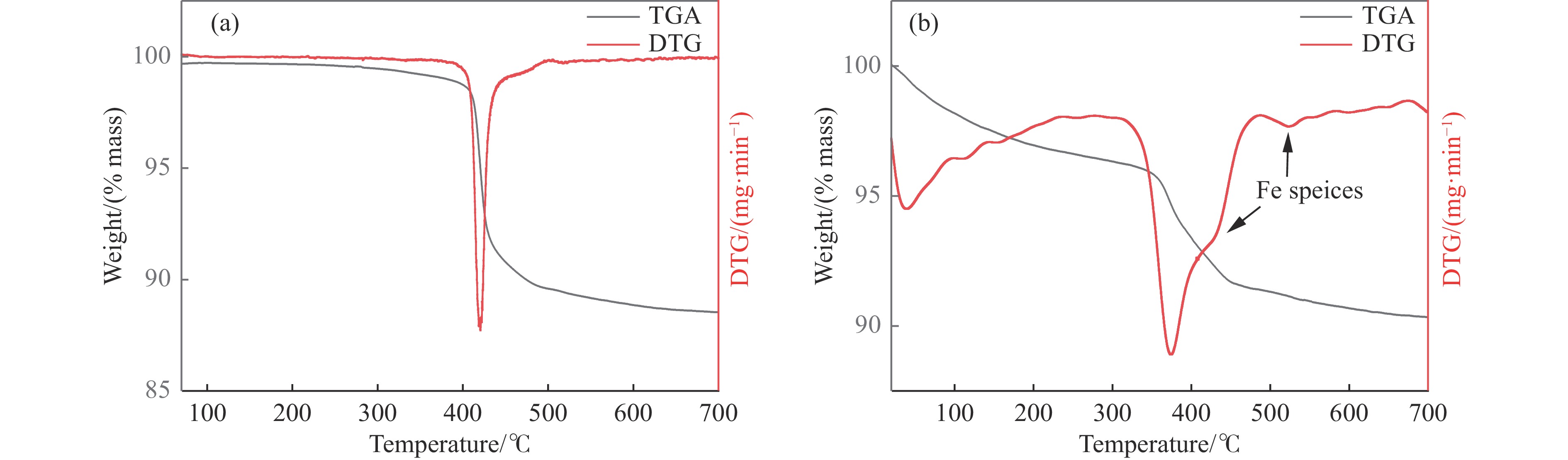
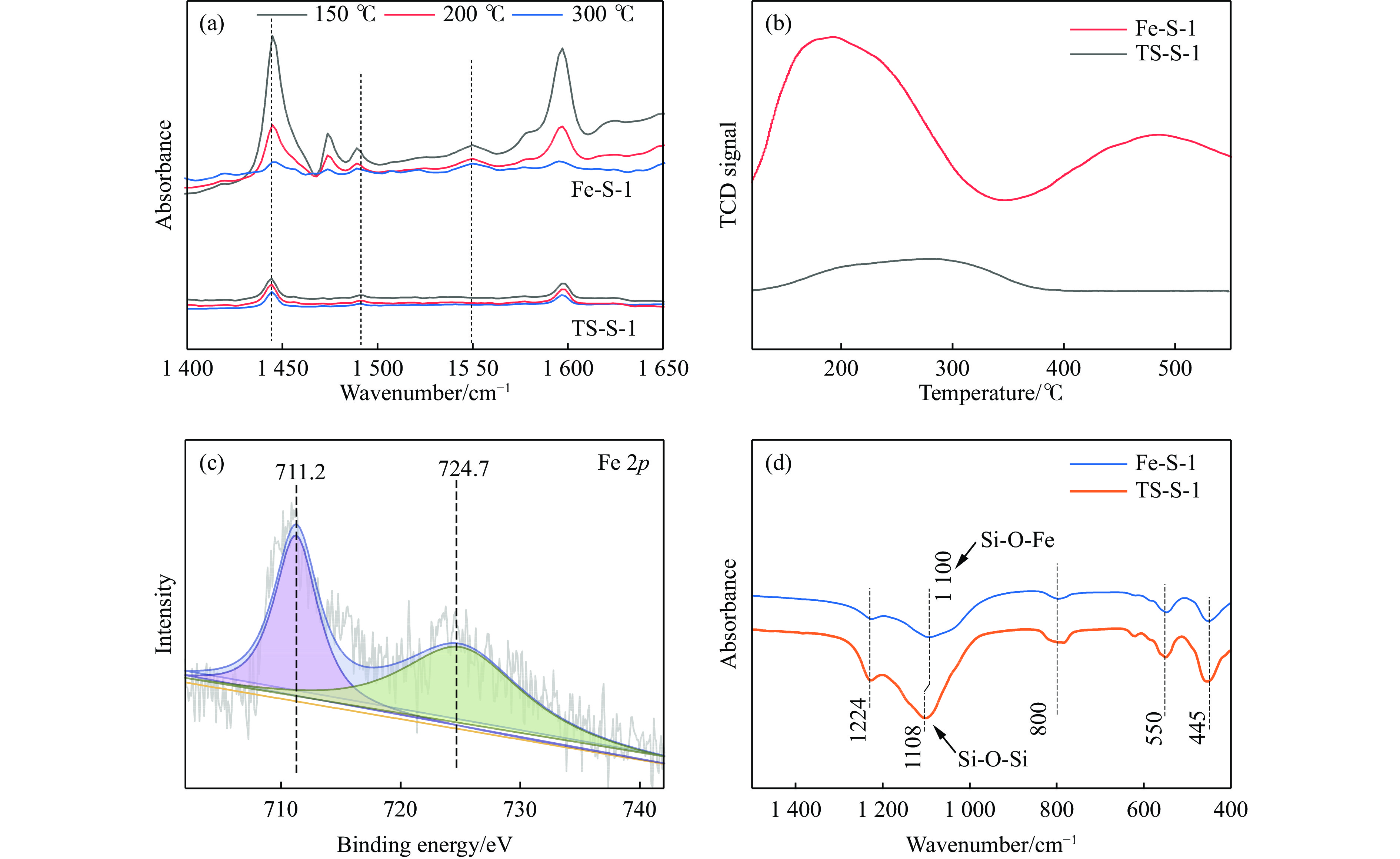
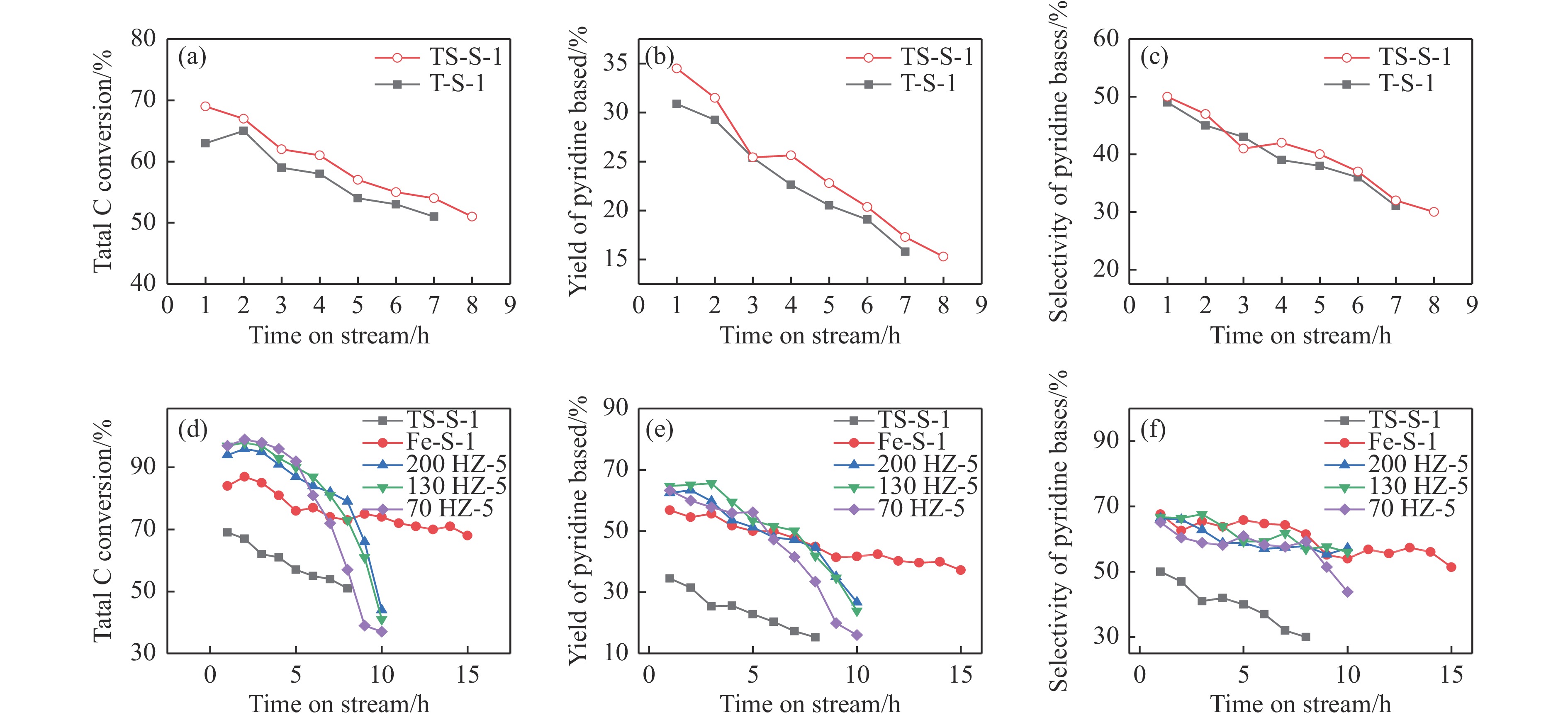
吡啶及其衍生物统称为吡啶碱,其广泛应用于农药、医药等领域。Chichibabin醛氨缩合反应是目前工业制取吡啶碱最广泛的路线。目前使用最广泛的ZSM-5分子筛受制于硅铝骨架结构的不稳定性,高活性反应周期较短(5 h),针对这一问题,本文选用热稳定性、水热稳定性优异的Silicalite-1分子筛,使用聚乙烯吡咯烷酮(PVP)作为胶体分散剂,在水热合成分子筛的过程中向骨架中引入Fe,结合XRD、SEM、TG、BET、NH3-TPD、Py-IR等表征方法探究了晶化条件对Silicalite-1分子筛结晶度、孔结构和酸性质的影响。实验结果表明,在晶种投入量15%、PVP添加量3.75%时产品相对结晶度达到最高(103%),粒径约为200 nm。改性后的Silicalite-1具有更丰富的酸位点,醛氨缩合反应的初始活性由66%增加至85%,在反应进行15 h后,原料转化率和吡啶碱收率分别保持在66%和40%以上。最后,本文提出的原位改性Silicalite-1分子筛策略极大扩宽了纯硅沸石在酸催化领域的应用,具有显著的科研价值和工业化潜力。
吡啶及其衍生物统称为吡啶碱,其广泛应用于农药、医药等领域。Chichibabin醛氨缩合反应是目前工业制取吡啶碱最广泛的路线。目前使用最广泛的ZSM-5分子筛受制于硅铝骨架结构的不稳定性,高活性反应周期较短(5 h),针对这一问题,本文选用热稳定性、水热稳定性优异的Silicalite-1分子筛,使用聚乙烯吡咯烷酮(PVP)作为胶体分散剂,在水热合成分子筛的过程中向骨架中引入Fe,结合XRD、SEM、TG、BET、NH3-TPD、Py-IR等表征方法探究了晶化条件对Silicalite-1分子筛结晶度、孔结构和酸性质的影响。实验结果表明,在晶种投入量15%、PVP添加量3.75%时产品相对结晶度达到最高(103%),粒径约为200 nm。改性后的Silicalite-1具有更丰富的酸位点,醛氨缩合反应的初始活性由66%增加至85%,在反应进行15 h后,原料转化率和吡啶碱收率分别保持在66%和40%以上。最后,本文提出的原位改性Silicalite-1分子筛策略极大扩宽了纯硅沸石在酸催化领域的应用,具有显著的科研价值和工业化潜力。
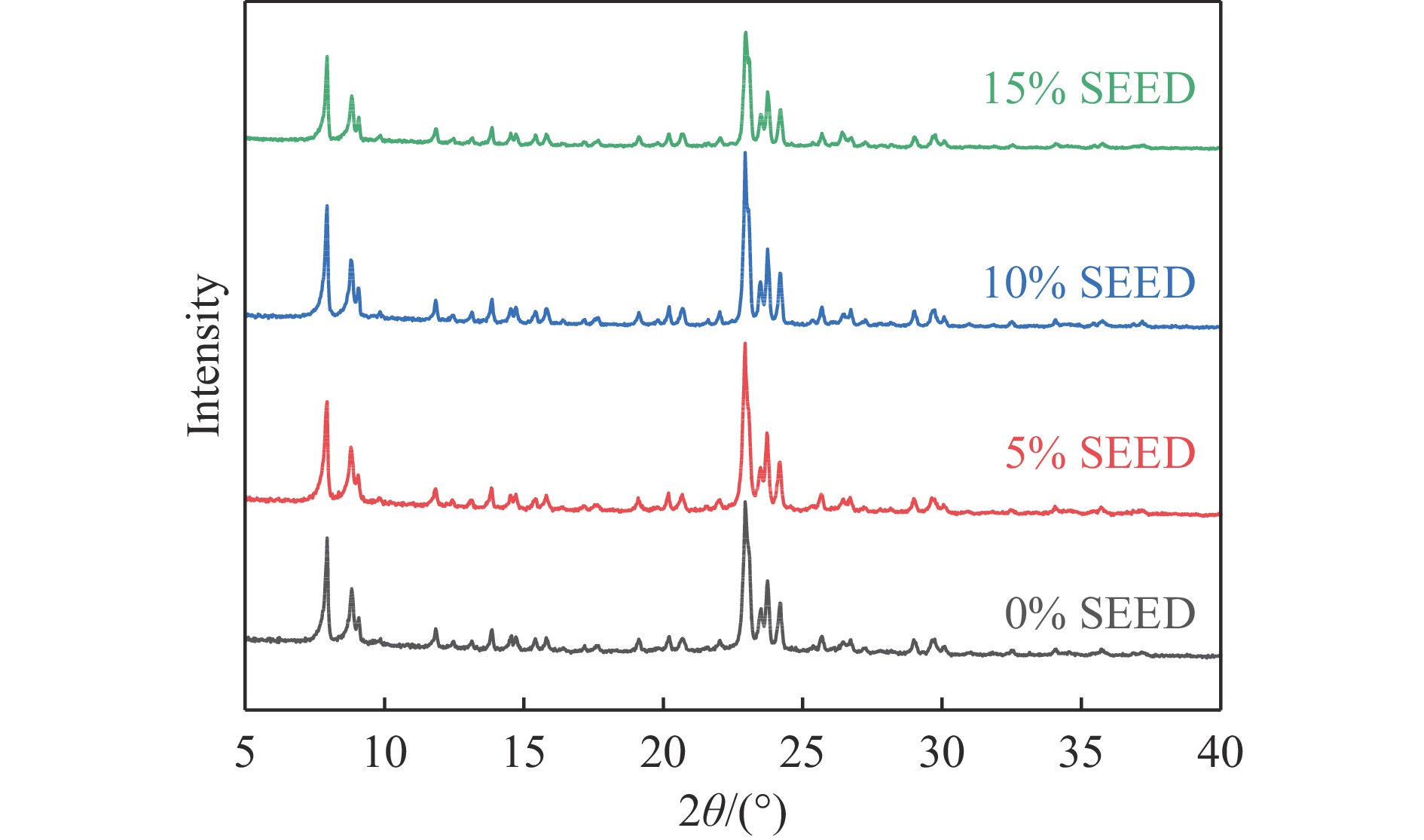

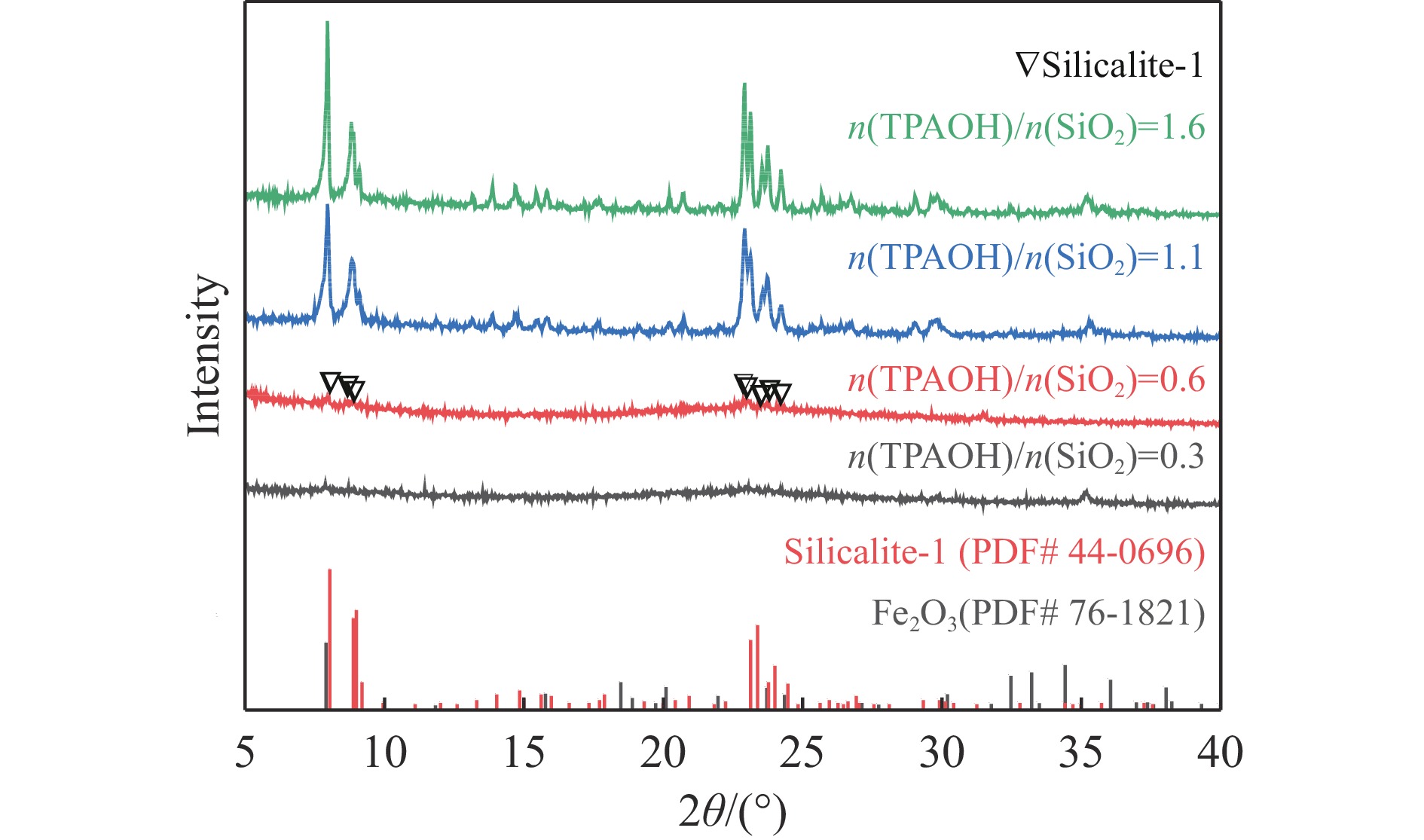



当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60439-1
摘要:
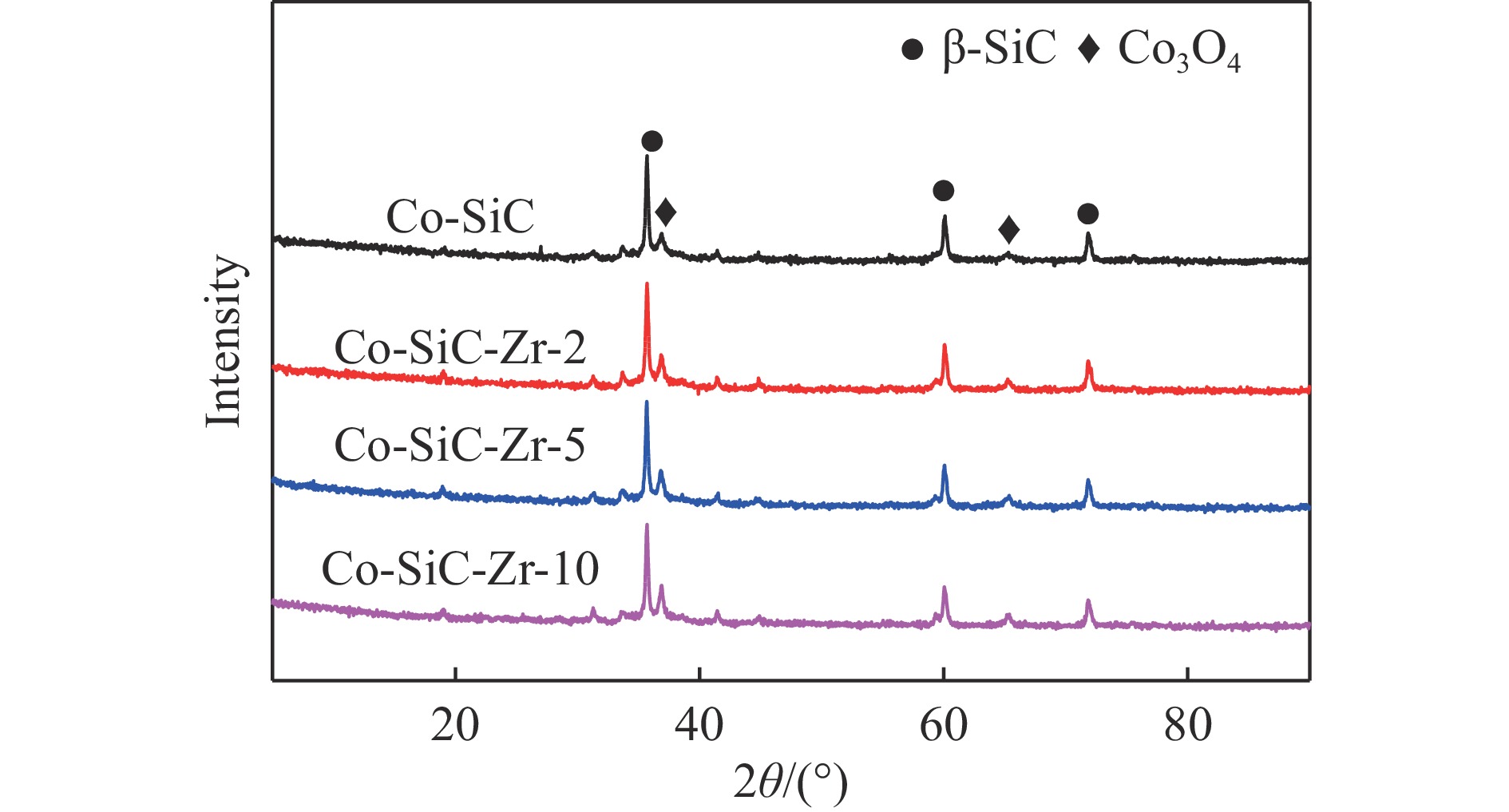
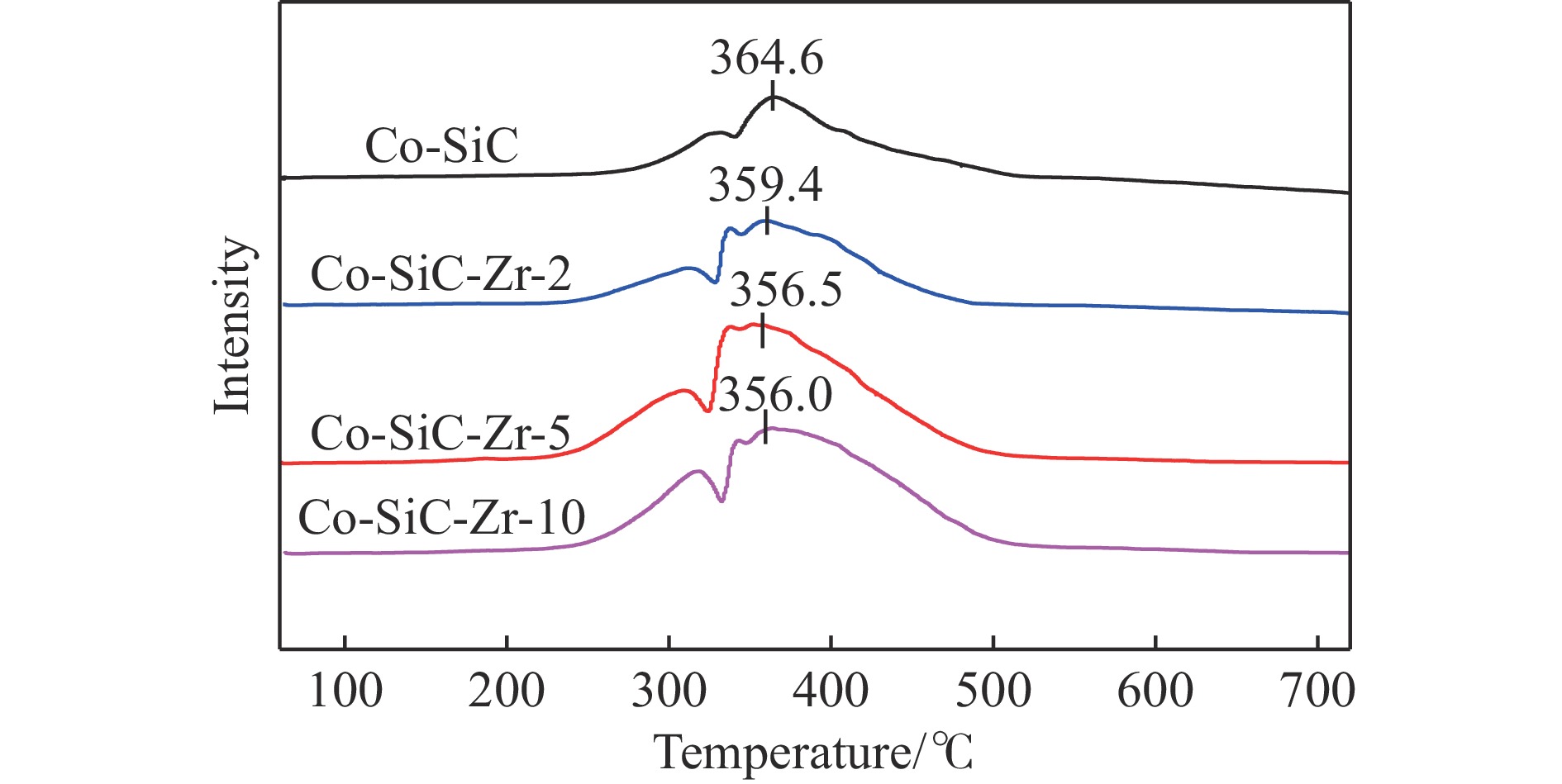
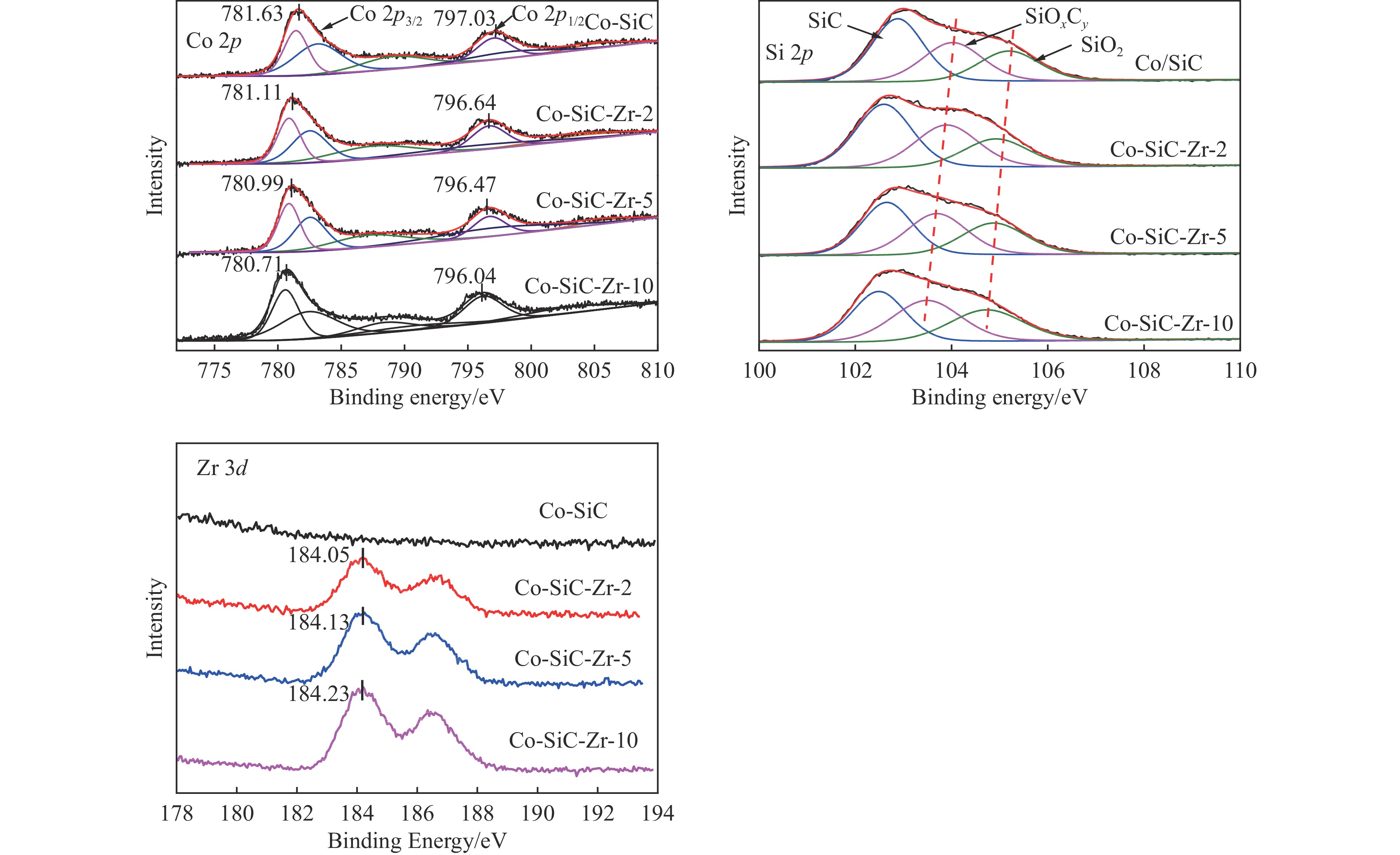
Co/SiC catalysts have exhibited excellent performance in Fischer-Tropsch synthesis reaction.,However, few research focuses on investigating the effect of SiC supports surface properties of on catalyst performance. In this study, ZrO2 was utilized to modify the SiC surface,leading to the preparation of a series of Co-ZrO2/SiC catalysts. The physicochemical properties of the catalyst were comprehensively analyzed by using N2 adsorption, XRD, H2-TPR, XPS analyses. Catalytic performance was evaluated using a fixed bed reactor, shedding light on the effect of ZrO2 modified SiC support on cobalt-based Fischer-Tropsch synthesis catalysts. The results indicated that ZrO2 surface modification on SiC resulted in an enhanced reduction degree of Co/SiC catalysts. Additionally, ZrO2 exhibited strong interaction with the amorphous phase on the SiC surface, thereby weakening the interaction between Co and the amorphous phase., This led to an increase in the electron density of cobalt species, consequently improving the selectivity of Co/SiC catalysts towards long-chain hydrocarbons.
Co/SiC catalysts have exhibited excellent performance in Fischer-Tropsch synthesis reaction.,However, few research focuses on investigating the effect of SiC supports surface properties of on catalyst performance. In this study, ZrO2 was utilized to modify the SiC surface,leading to the preparation of a series of Co-ZrO2/SiC catalysts. The physicochemical properties of the catalyst were comprehensively analyzed by using N2 adsorption, XRD, H2-TPR, XPS analyses. Catalytic performance was evaluated using a fixed bed reactor, shedding light on the effect of ZrO2 modified SiC support on cobalt-based Fischer-Tropsch synthesis catalysts. The results indicated that ZrO2 surface modification on SiC resulted in an enhanced reduction degree of Co/SiC catalysts. Additionally, ZrO2 exhibited strong interaction with the amorphous phase on the SiC surface, thereby weakening the interaction between Co and the amorphous phase., This led to an increase in the electron density of cobalt species, consequently improving the selectivity of Co/SiC catalysts towards long-chain hydrocarbons.
当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60440-8
摘要:
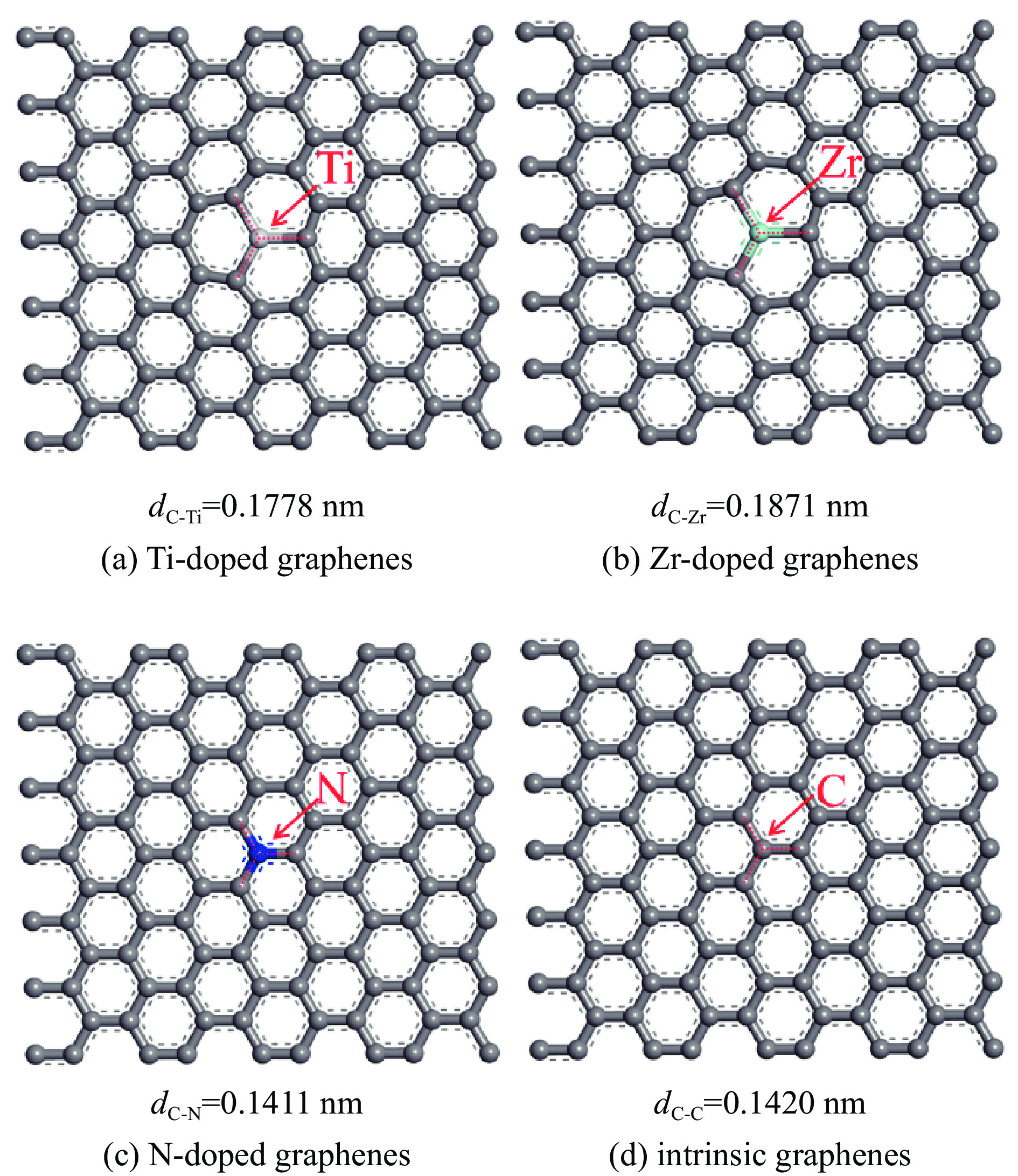
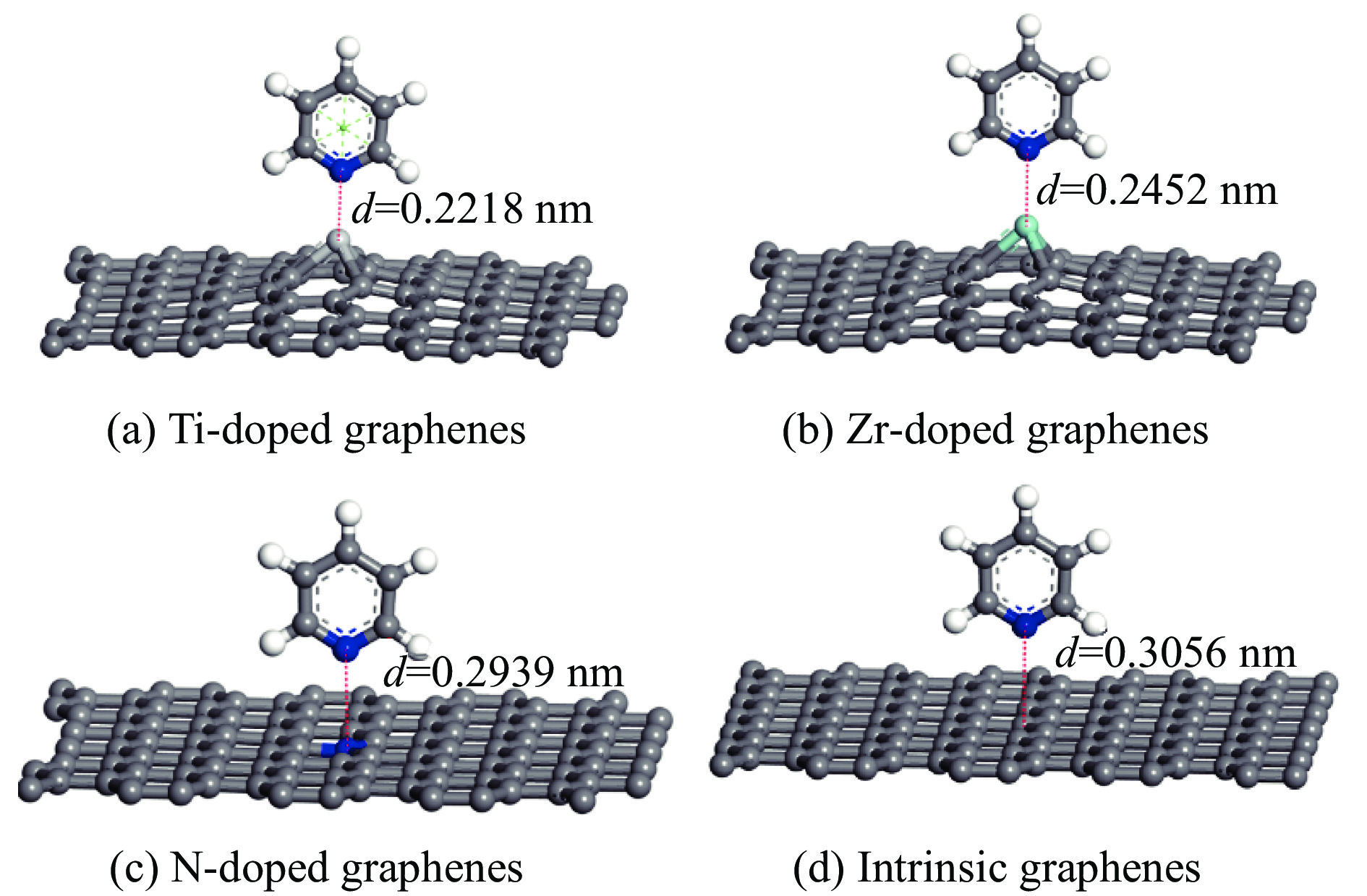
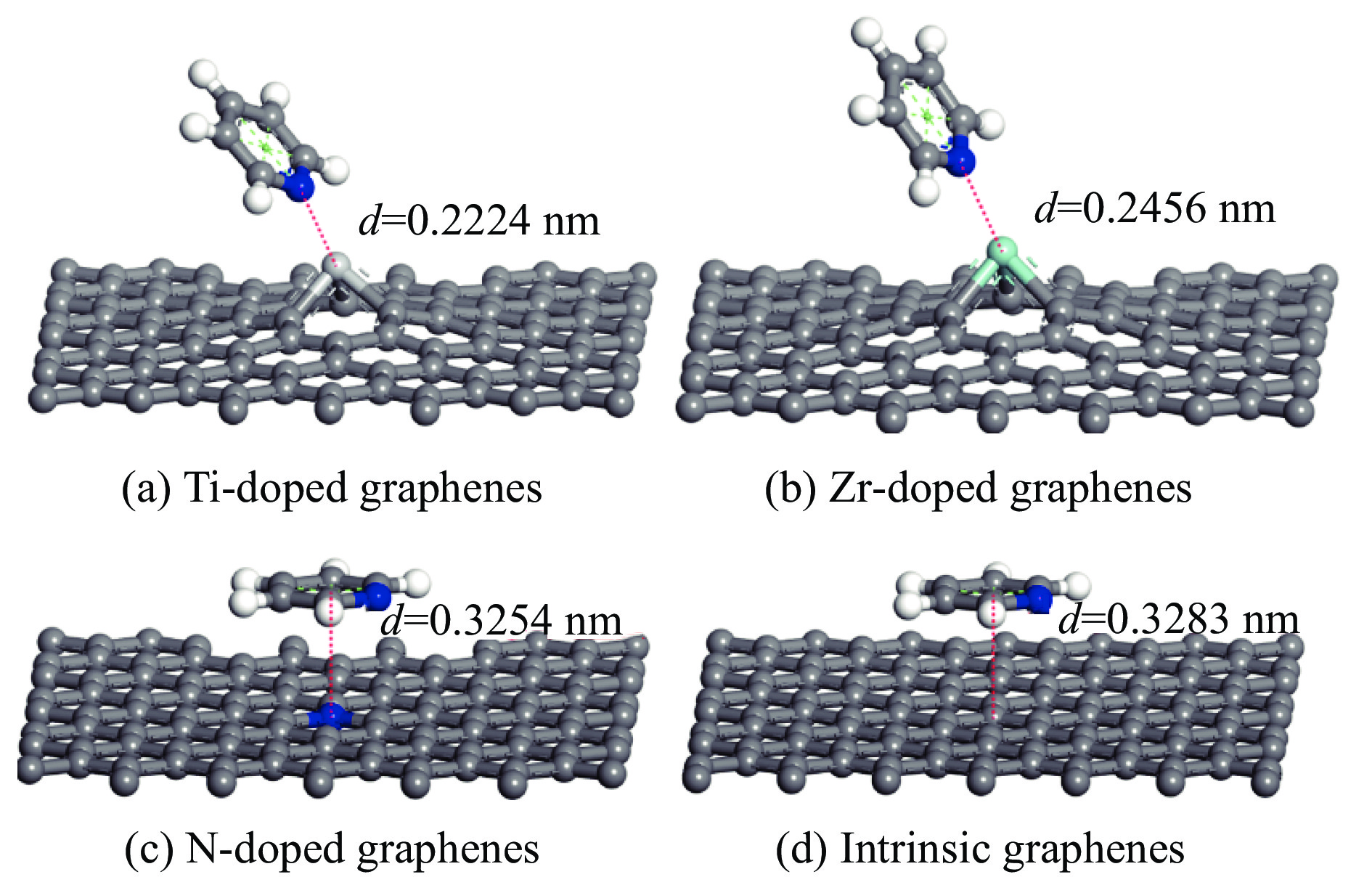
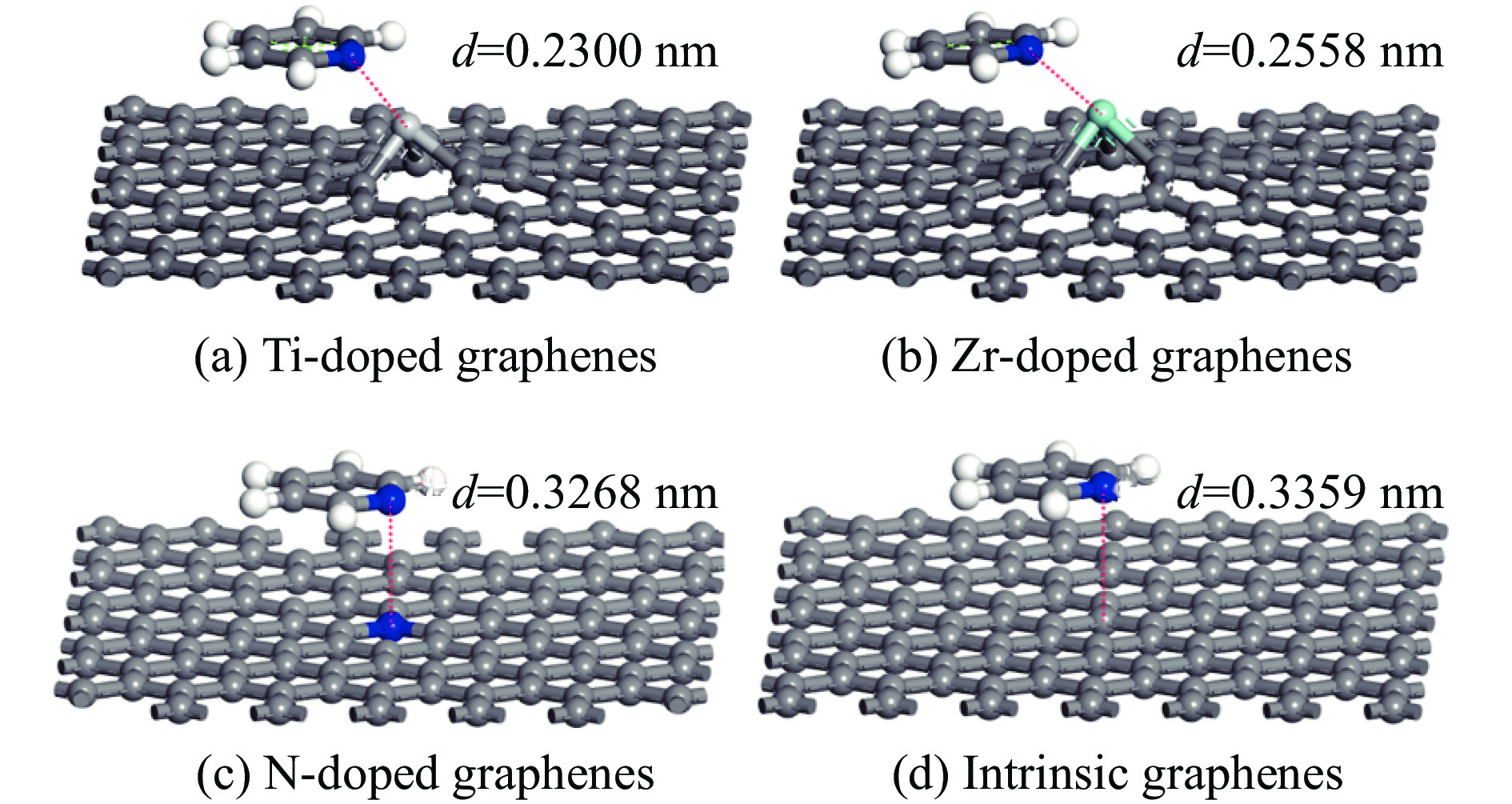
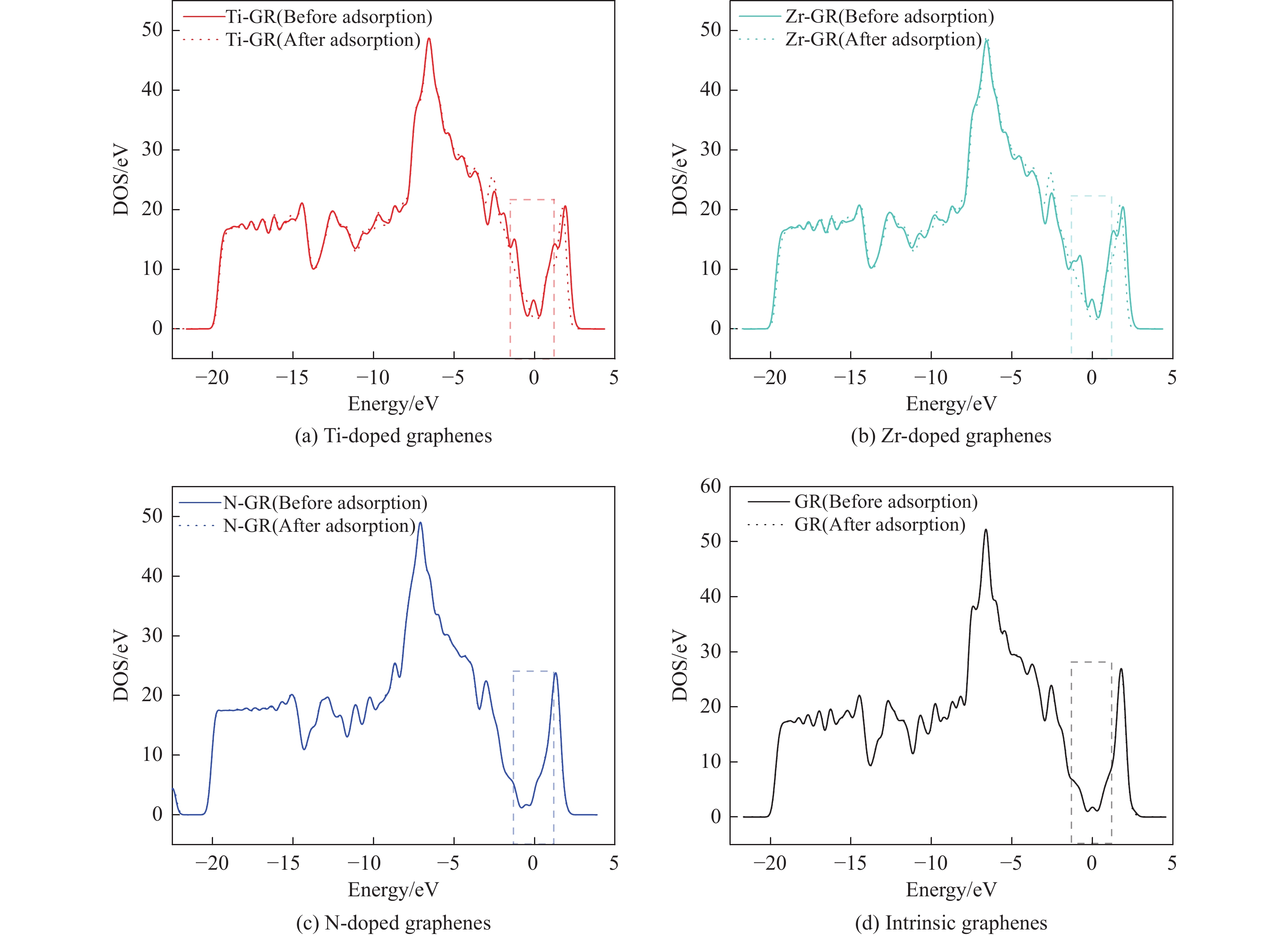
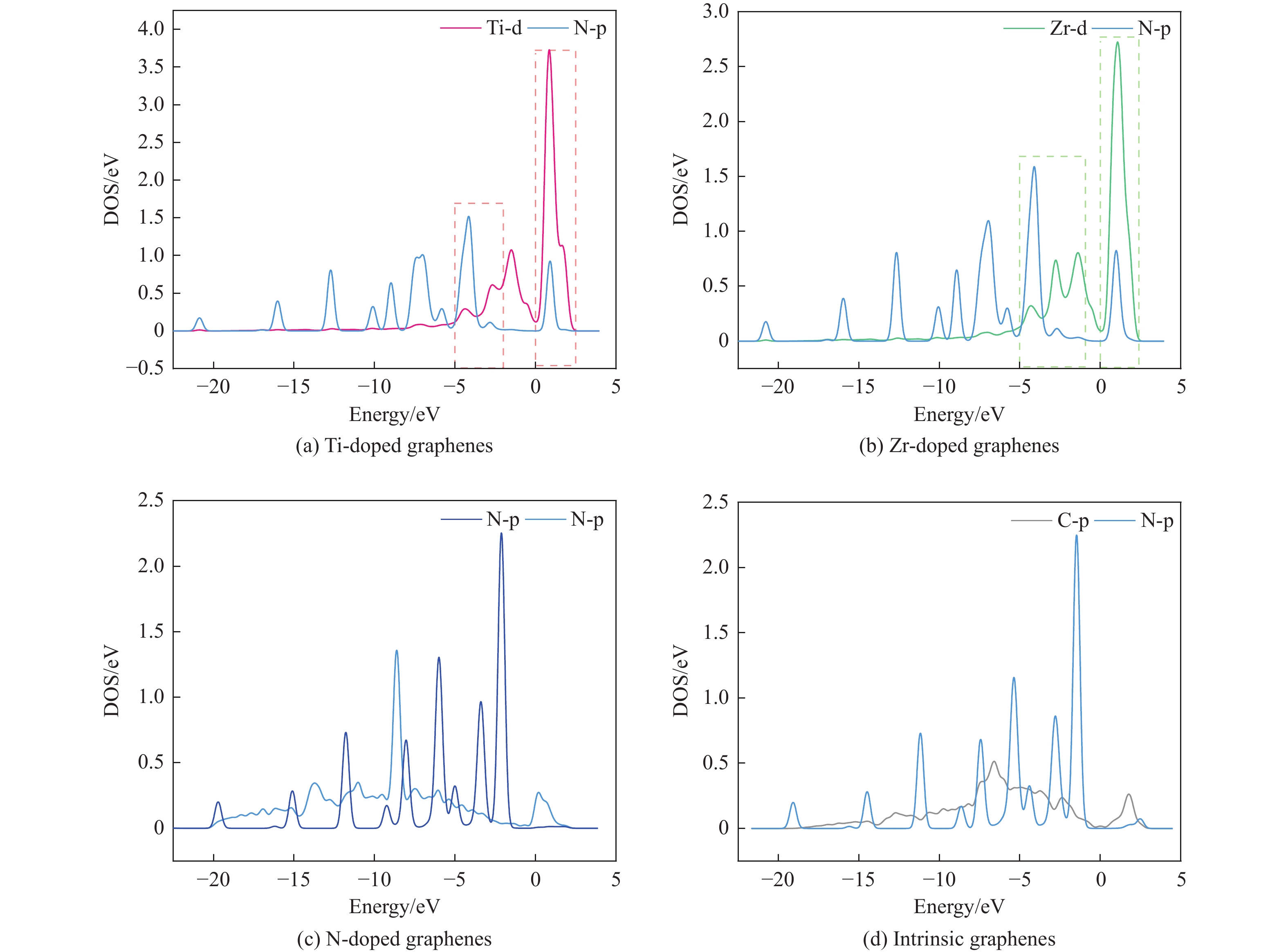
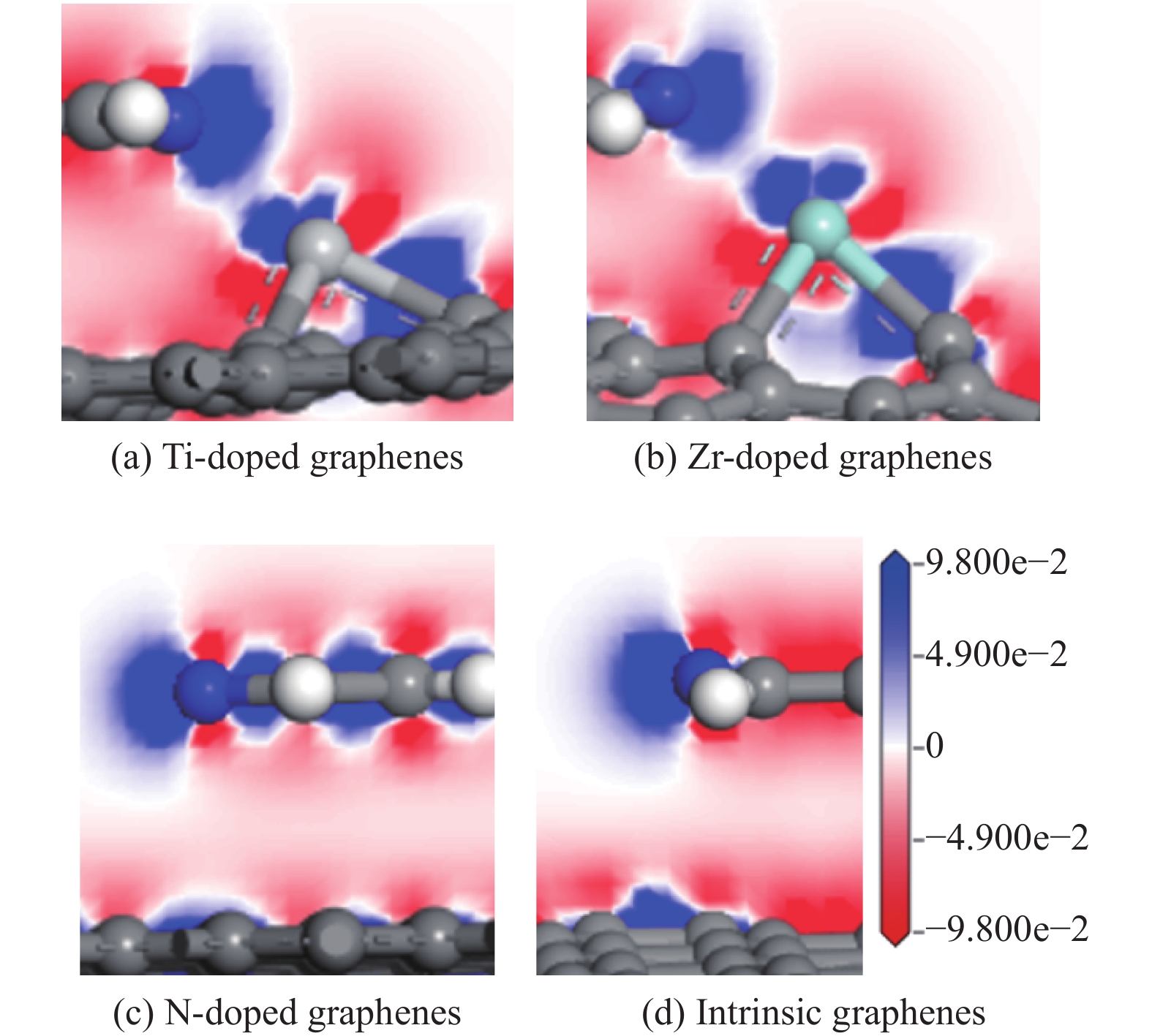
采用密度泛函方法,研究了Ti、Zr和N掺杂及本征石墨烯对柴油中典型碱性氮化物吡啶的吸附行为,讨论了相应的吸附能、吸附构型、马利肯电荷转移、差分电荷密度和态密度。结果表明,金属Ti、Zr掺杂能显著增强吡啶在石墨烯表面的吸附能,非金属N掺杂可略微增加吡啶和石墨烯表面间的吸附能。吡啶在不同原子修饰的石墨烯表面的吸附能大小顺序为Ti掺杂石墨烯>Zr掺杂石墨烯>N掺杂石墨烯>本征石墨烯,吡啶可与Ti、Zr掺杂石墨烯发生N-Ti、N-Zr和π-π作用,与N掺杂石墨烯、本征石墨烯发生N-N、C-N和π-π作用。进一步分析发现,吡啶和金属Ti、Zr掺杂石墨烯表面存在明显的电子转移和化学键的形成,而和非金属N掺杂石墨烯及本征石墨烯间并无化学键形成。吡啶与Ti、Zr掺杂石墨烯发生化学吸附,与N掺杂石墨烯、本征石墨烯发生物理吸附。吡啶更稳定的吸附在Ti、Zr掺杂石墨烯表面。
采用密度泛函方法,研究了Ti、Zr和N掺杂及本征石墨烯对柴油中典型碱性氮化物吡啶的吸附行为,讨论了相应的吸附能、吸附构型、马利肯电荷转移、差分电荷密度和态密度。结果表明,金属Ti、Zr掺杂能显著增强吡啶在石墨烯表面的吸附能,非金属N掺杂可略微增加吡啶和石墨烯表面间的吸附能。吡啶在不同原子修饰的石墨烯表面的吸附能大小顺序为Ti掺杂石墨烯>Zr掺杂石墨烯>N掺杂石墨烯>本征石墨烯,吡啶可与Ti、Zr掺杂石墨烯发生N-Ti、N-Zr和π-π作用,与N掺杂石墨烯、本征石墨烯发生N-N、C-N和π-π作用。进一步分析发现,吡啶和金属Ti、Zr掺杂石墨烯表面存在明显的电子转移和化学键的形成,而和非金属N掺杂石墨烯及本征石墨烯间并无化学键形成。吡啶与Ti、Zr掺杂石墨烯发生化学吸附,与N掺杂石墨烯、本征石墨烯发生物理吸附。吡啶更稳定的吸附在Ti、Zr掺杂石墨烯表面。


当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024005
摘要:
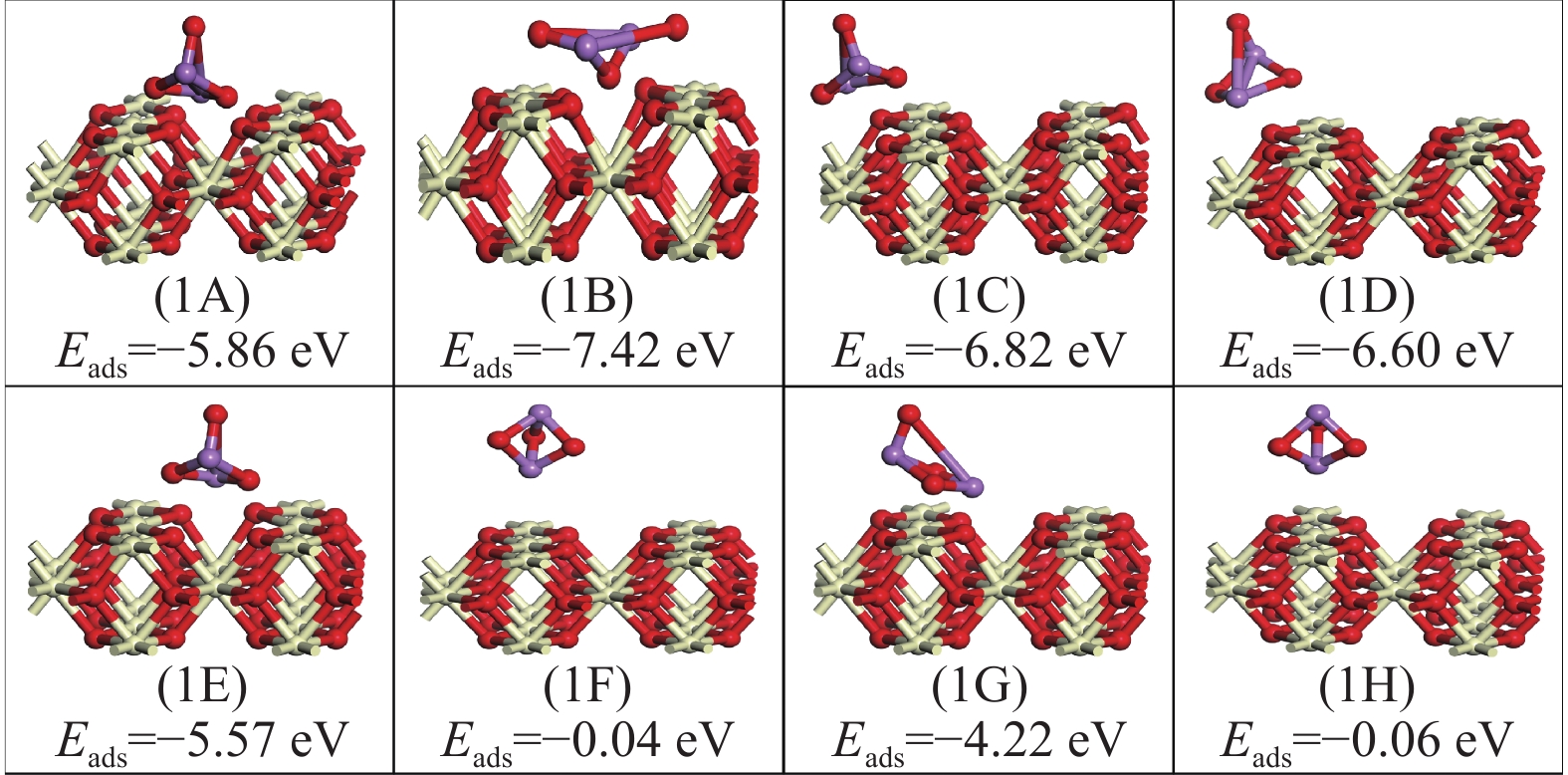
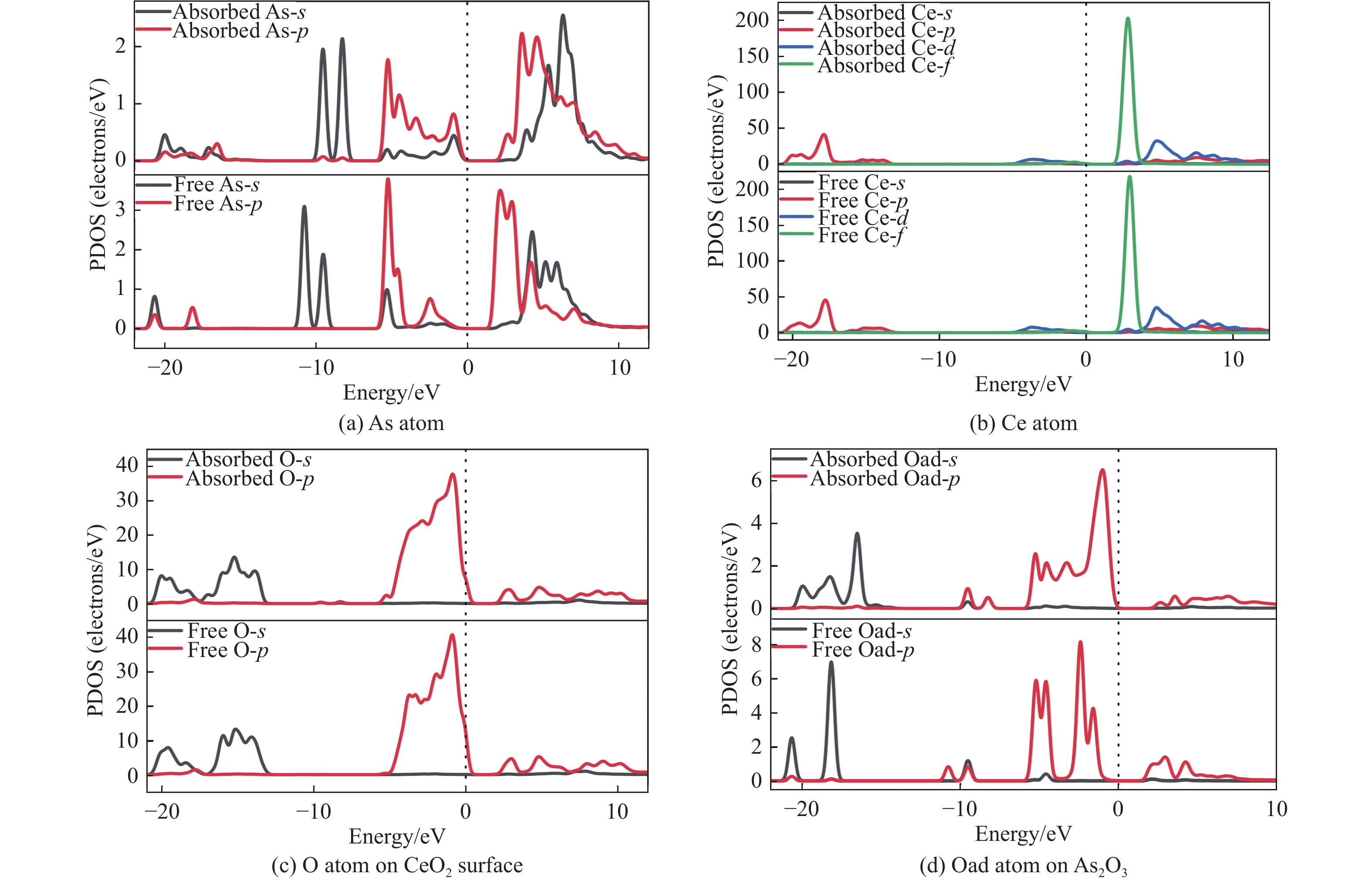
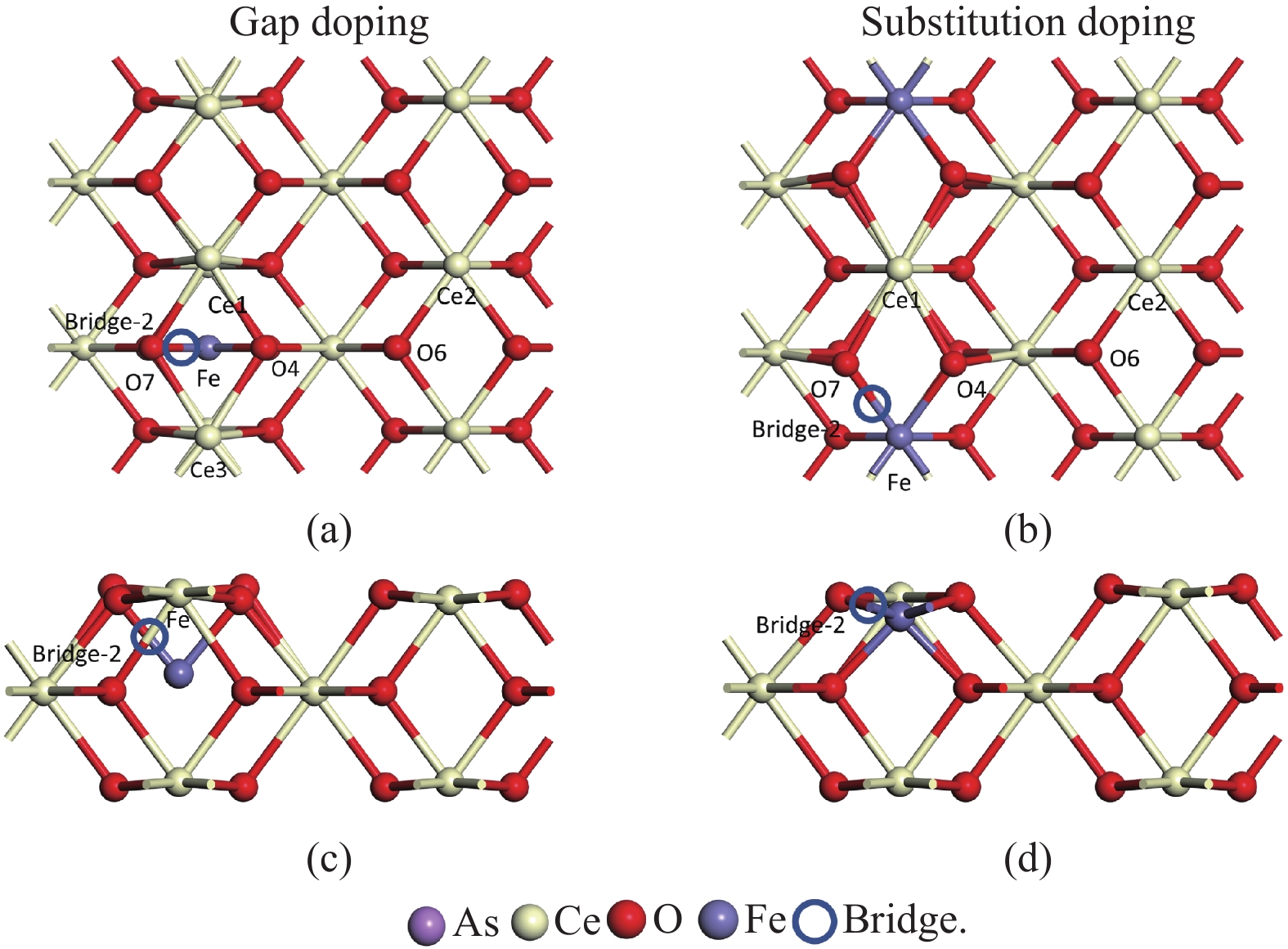
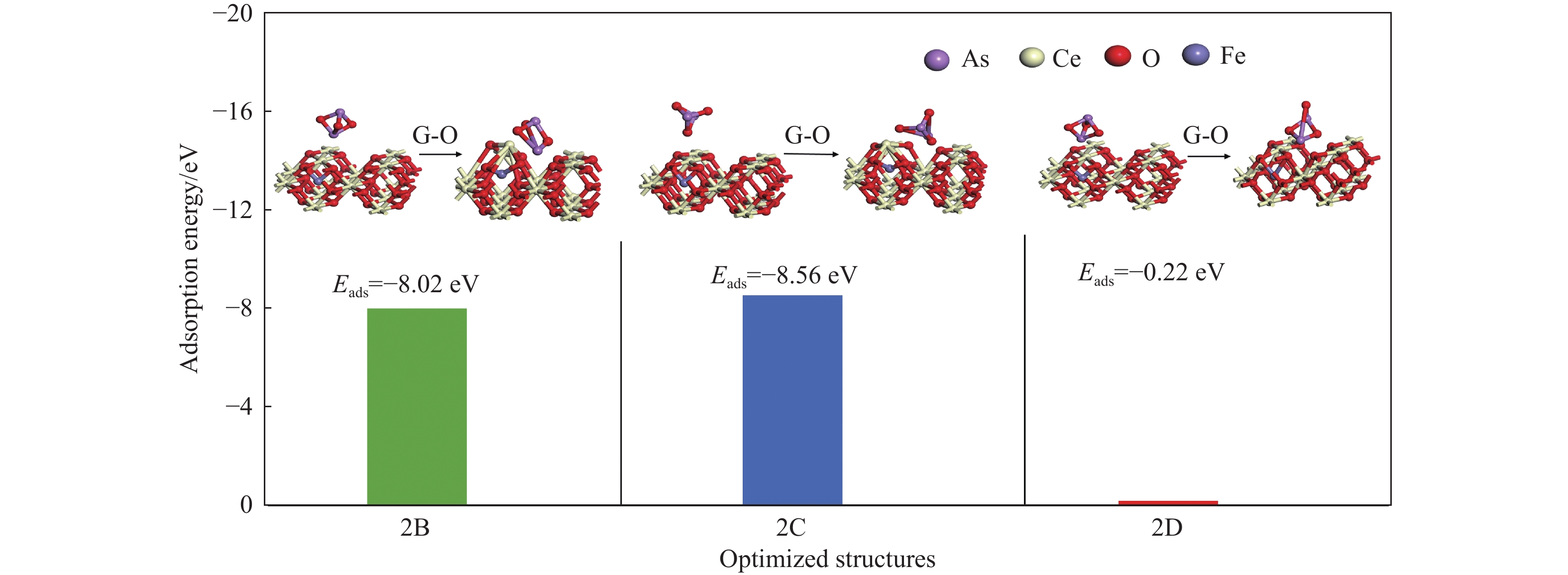
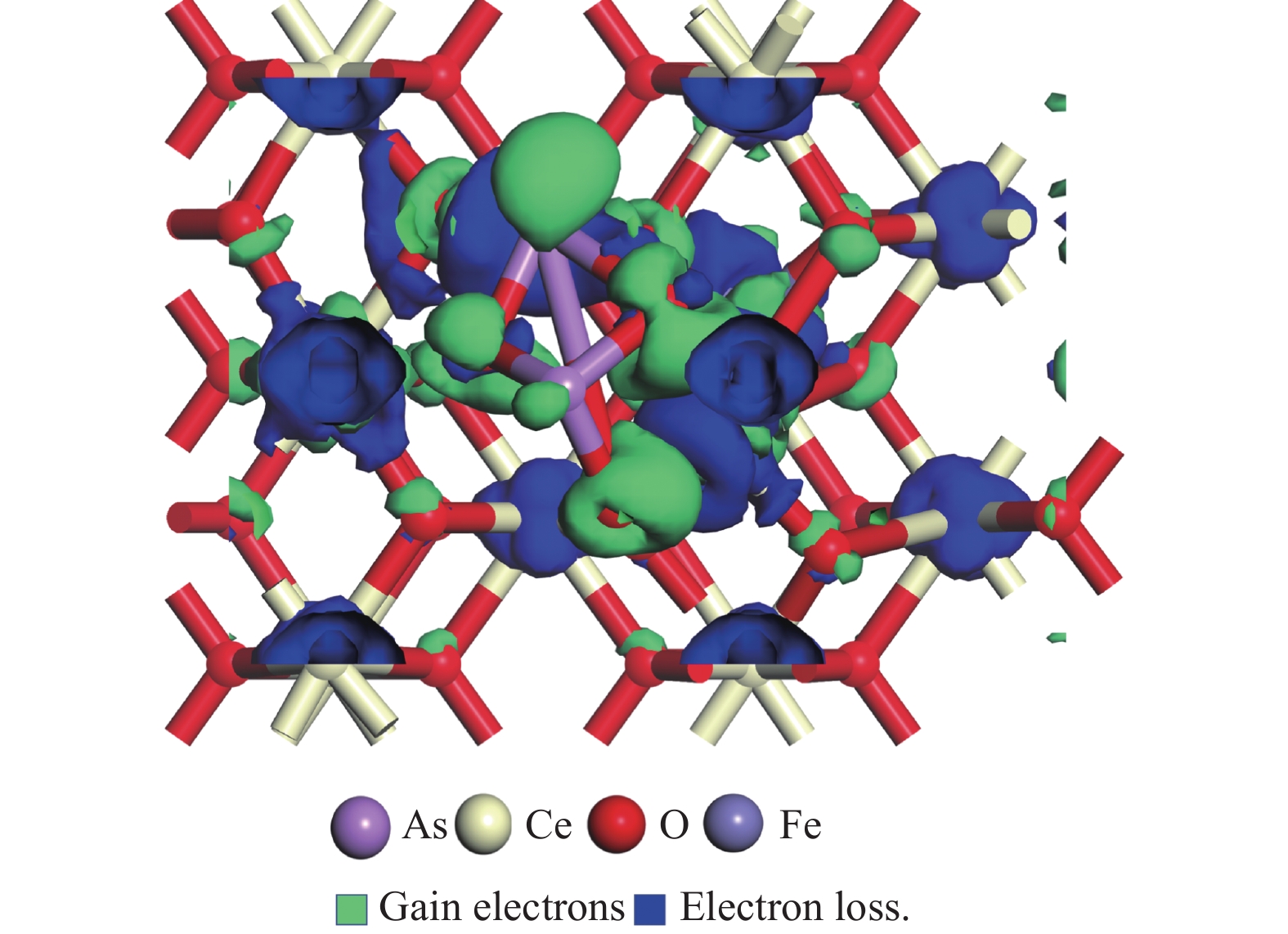
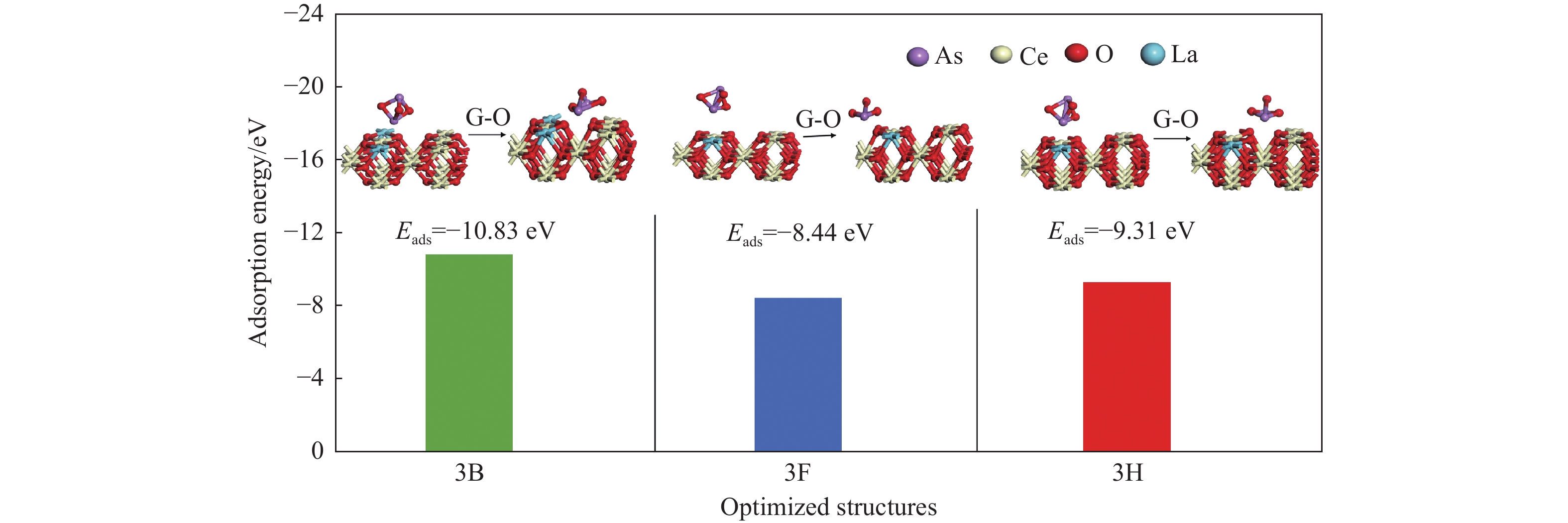
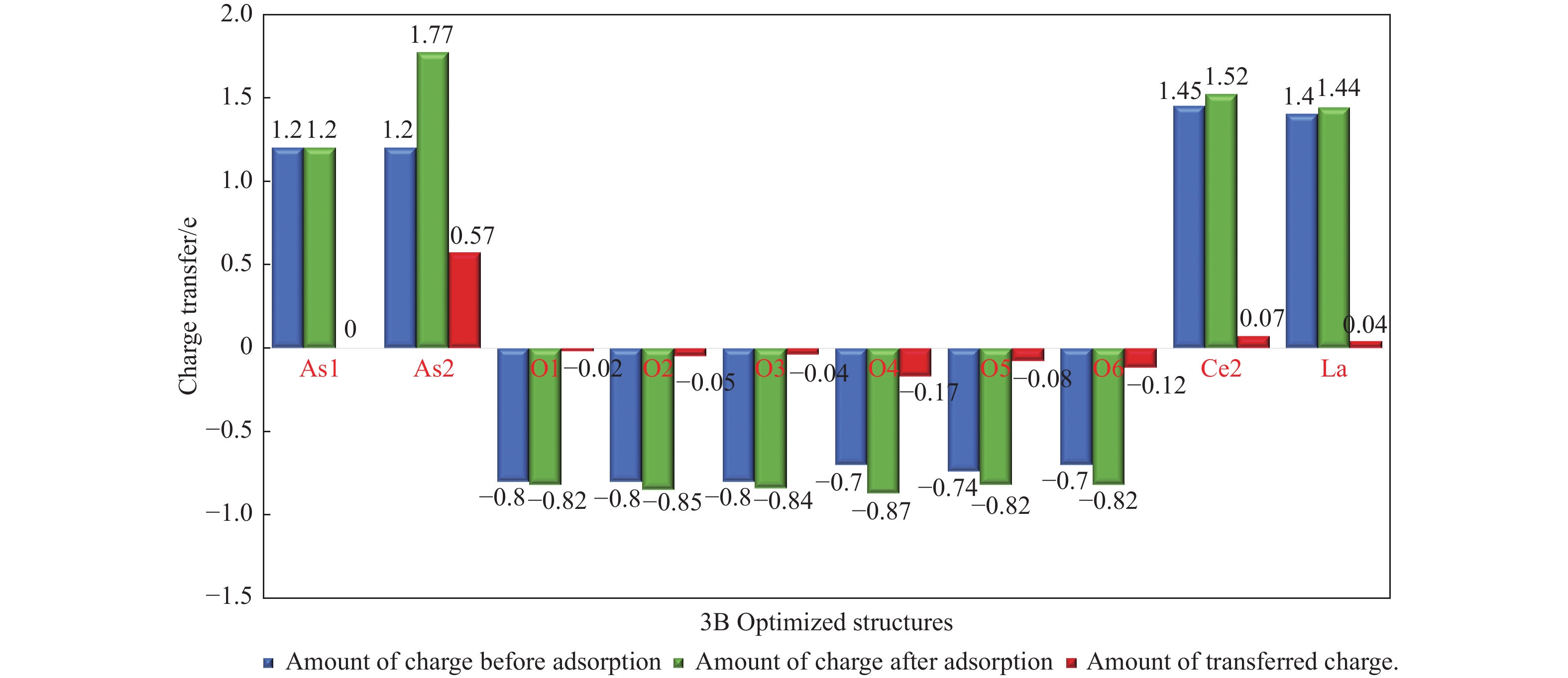
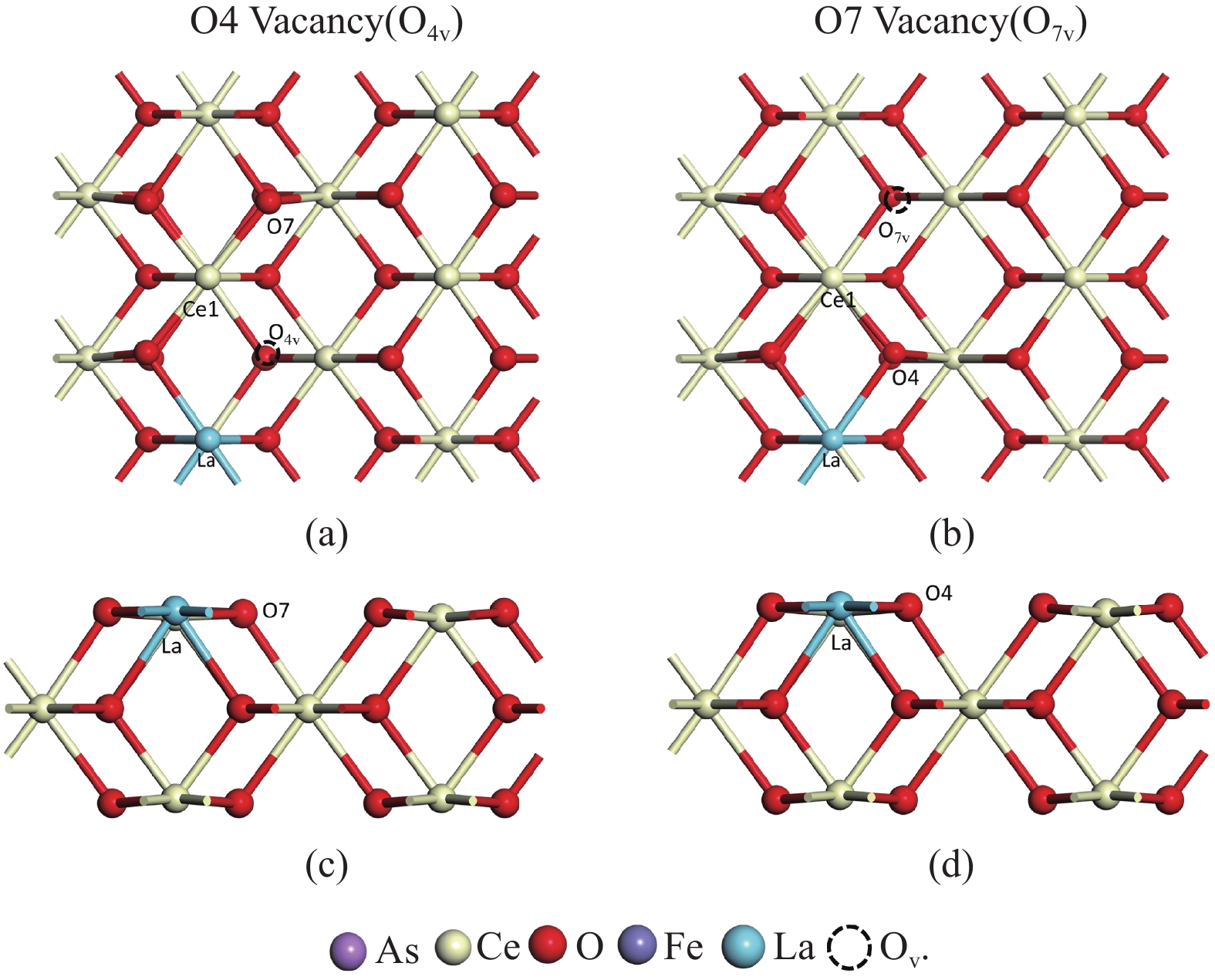
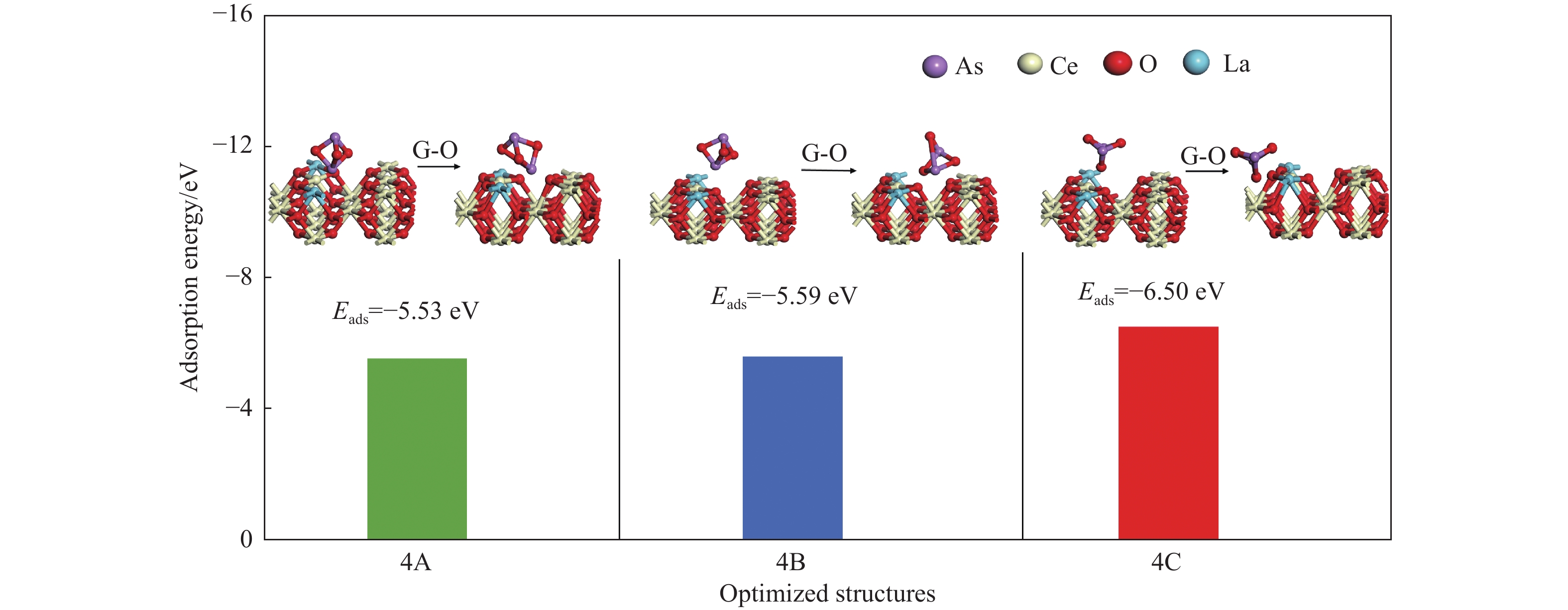
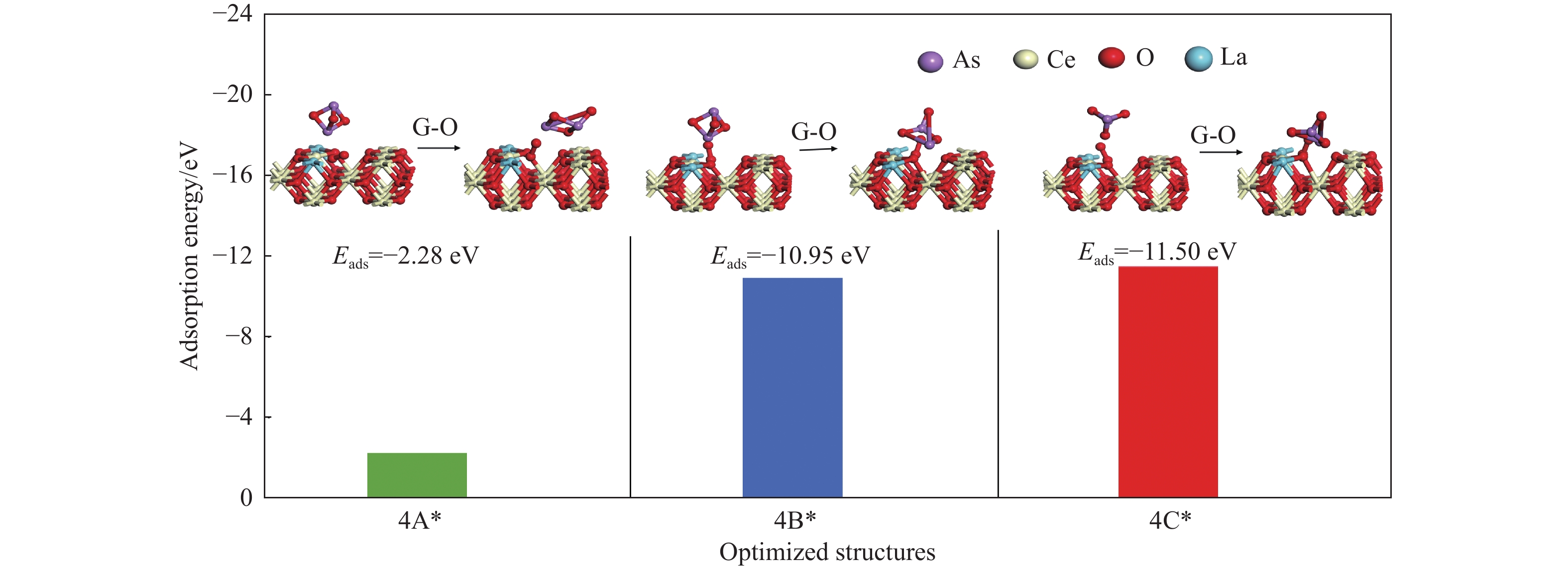
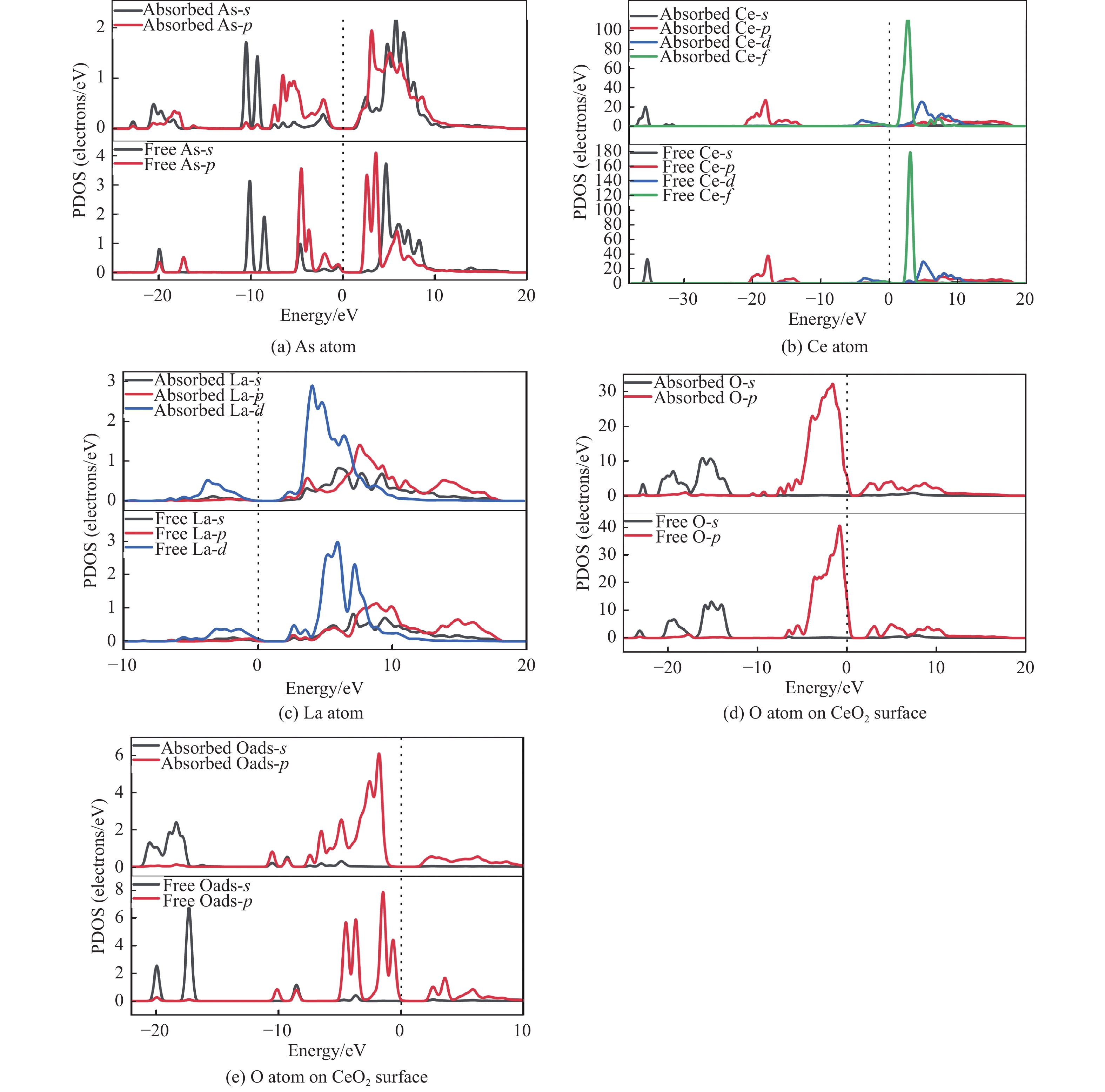
采用密度泛函理论研究了As2O3(g)在Fe、La掺杂CeO2(110)表面及氧缺陷LaCeO(110)表面的吸附行为,探索了LaCeO表面砷吸附能力显著高于FeCeO表面的主要原因。结果表明,As2O3(g)的吸附效果与吸附位点数量、吸附能、键长和电荷转移密切相关。纯CeO2表面的吸附主要为化学吸附,吸附能绝对值大于−4.22 eV,电荷转移量为−0.19− −0.31 e,As2O3得到电荷带负电,起表面受主作用,因此吸附量较小。FeCeO(110)表面新增Fe顶位和Bridge-2桥位两个吸附位,其中,Fe顶位为化学吸附,Fe掺杂改变了FeCeO表面电子分布和晶格结构,但并未改变As2O3与FeCeO之间的电荷转移方向,因此,As2O3仍呈负离子形式吸附。LaCeO(110)表面新增了三个吸附位:La顶位、Bridge-3桥位和Hollow-2空位,La掺杂改变了As2O3与LaCeO之间的电荷转移方向,使得As2O3失电子呈正离子吸附,起表面施主作用,因此,吸附能力增强。无O2环境下,单一O缺陷LaCeO(110)表面吸附能力低于完整LaCeO表面;有O2环境下,O缺陷有利于As2O3的吸附。
采用密度泛函理论研究了As2O3(g)在Fe、La掺杂CeO2(110)表面及氧缺陷LaCeO(110)表面的吸附行为,探索了LaCeO表面砷吸附能力显著高于FeCeO表面的主要原因。结果表明,As2O3(g)的吸附效果与吸附位点数量、吸附能、键长和电荷转移密切相关。纯CeO2表面的吸附主要为化学吸附,吸附能绝对值大于−4.22 eV,电荷转移量为−0.19− −0.31 e,As2O3得到电荷带负电,起表面受主作用,因此吸附量较小。FeCeO(110)表面新增Fe顶位和Bridge-2桥位两个吸附位,其中,Fe顶位为化学吸附,Fe掺杂改变了FeCeO表面电子分布和晶格结构,但并未改变As2O3与FeCeO之间的电荷转移方向,因此,As2O3仍呈负离子形式吸附。LaCeO(110)表面新增了三个吸附位:La顶位、Bridge-3桥位和Hollow-2空位,La掺杂改变了As2O3与LaCeO之间的电荷转移方向,使得As2O3失电子呈正离子吸附,起表面施主作用,因此,吸附能力增强。无O2环境下,单一O缺陷LaCeO(110)表面吸附能力低于完整LaCeO表面;有O2环境下,O缺陷有利于As2O3的吸附。



当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024007
摘要:
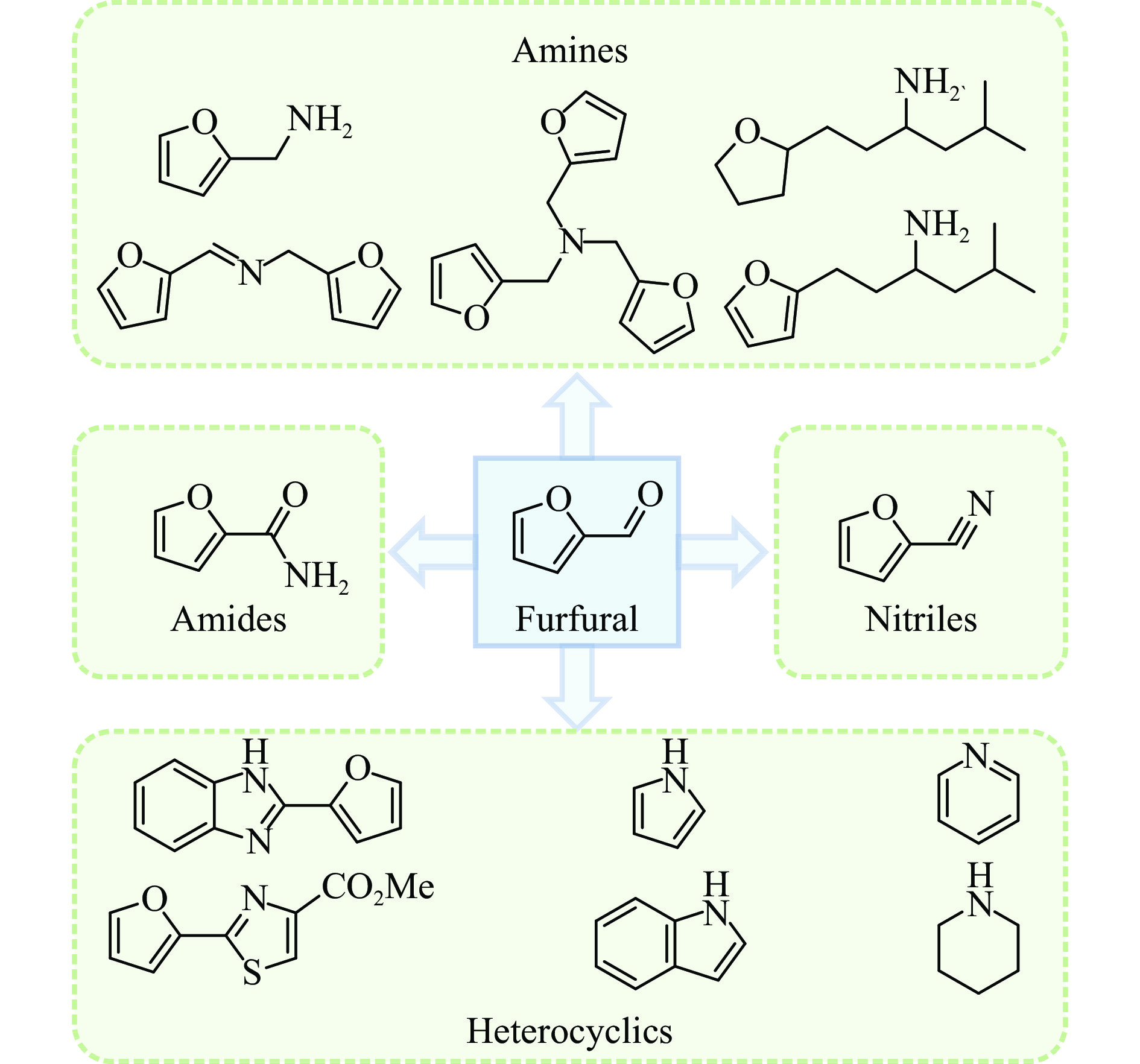
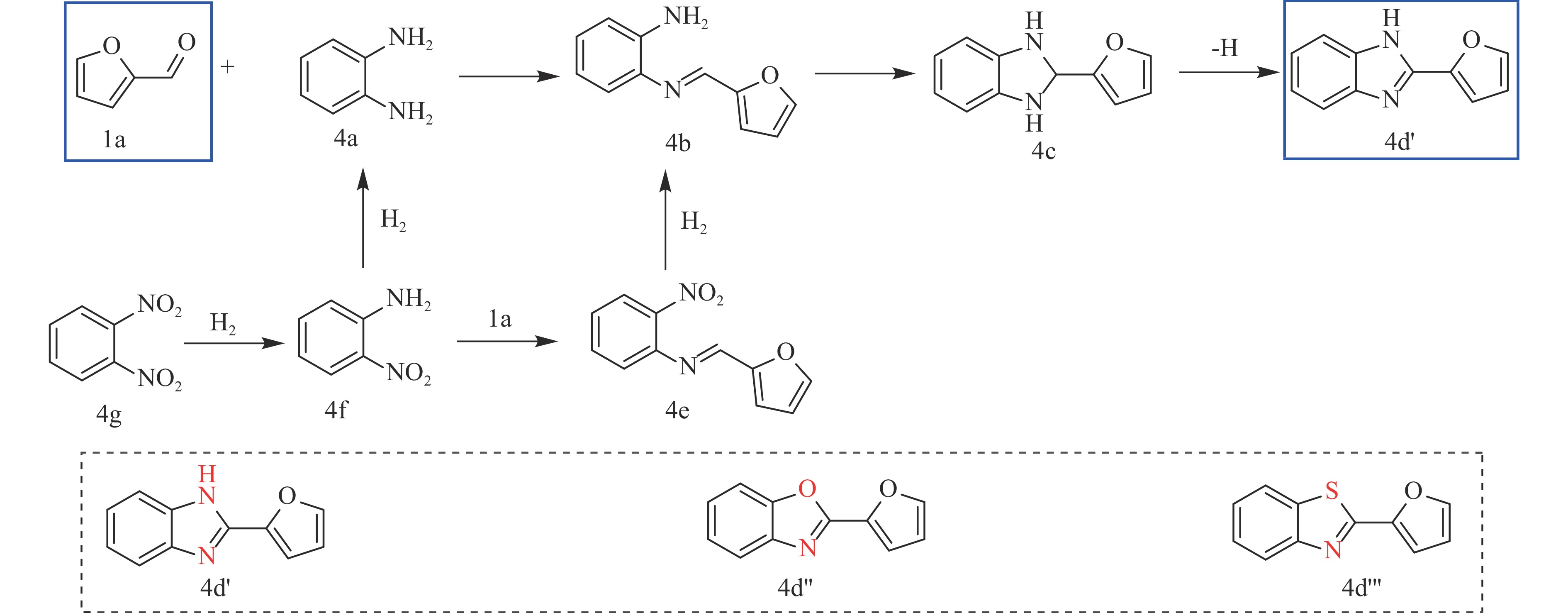
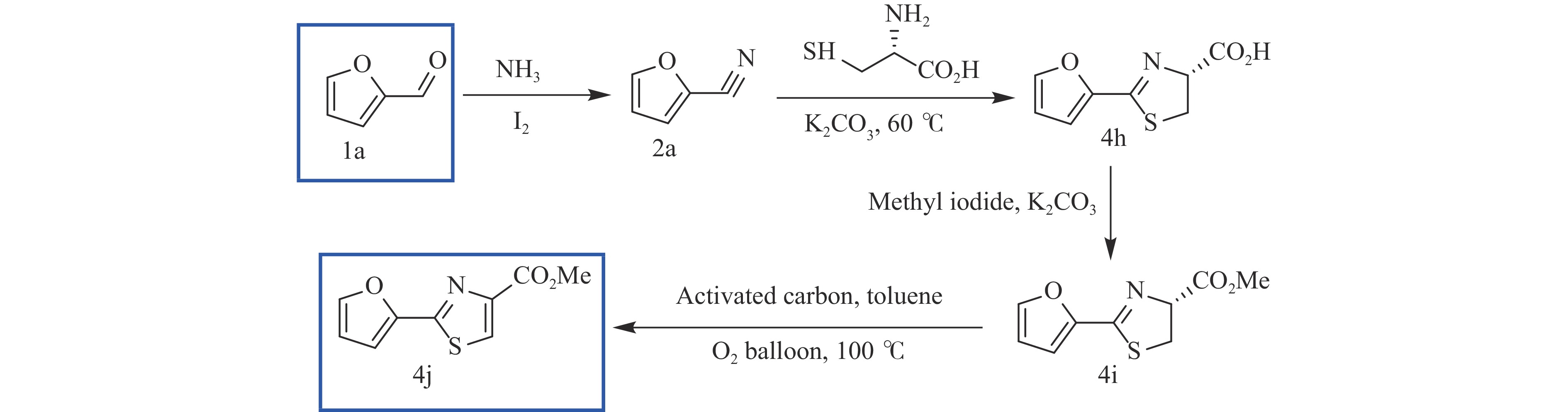
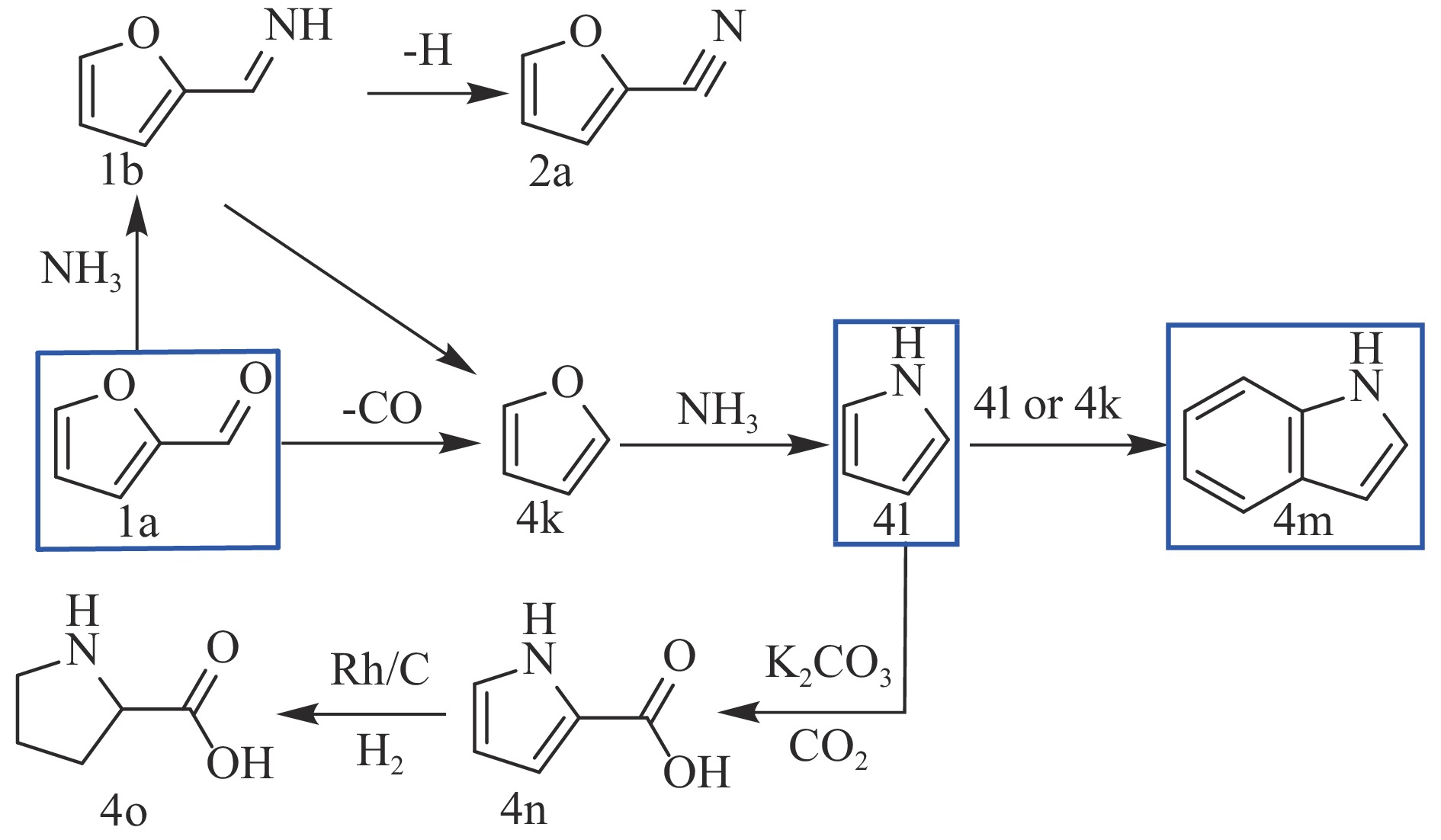
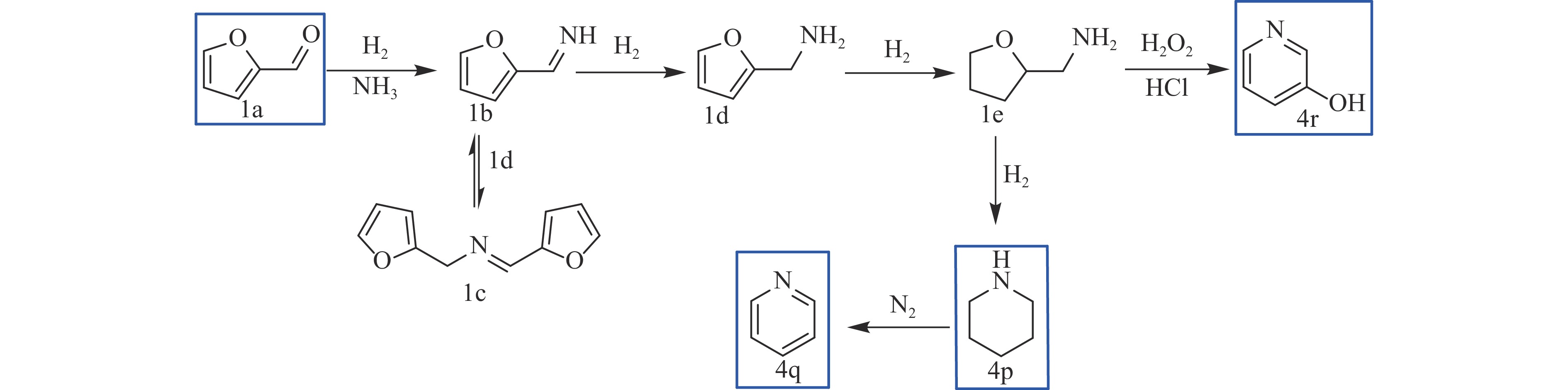
糠醛作为最有潜力的生物质基平台化合物之一,可通过化学催化转化为一系列高附加值的化学品和燃料。其中,含氮化合物具有广泛的生物活性,常用于合成药物分子和生物塑料等功能性材料。糠醛通过还原胺化、氨氧化、氧化偶联等过程,可以合成不同类型的含氮化合物,具有巨大的研究前景和应用潜力。本论文综述了近年来以糠醛为原料合成各种高值含氮化合物的研究进展,包括胺类化合物(伯胺、仲胺和叔胺)、腈类化合物、酰胺类化合物和杂环类化合物(苯并杂环类、噻唑类、吡咯、吲哚、哌啶和吡啶等)。重点关注合成方法、催化剂类型、反应路径和反应机理,同时分析了催化剂和氮源对产物分布的影响。该综述为今后生物质基糠醛转化为含氮化合物的研究提供了一些基础信息,为发展更多高效的糠醛催化转化体系提供依据和系统性知识。
糠醛作为最有潜力的生物质基平台化合物之一,可通过化学催化转化为一系列高附加值的化学品和燃料。其中,含氮化合物具有广泛的生物活性,常用于合成药物分子和生物塑料等功能性材料。糠醛通过还原胺化、氨氧化、氧化偶联等过程,可以合成不同类型的含氮化合物,具有巨大的研究前景和应用潜力。本论文综述了近年来以糠醛为原料合成各种高值含氮化合物的研究进展,包括胺类化合物(伯胺、仲胺和叔胺)、腈类化合物、酰胺类化合物和杂环类化合物(苯并杂环类、噻唑类、吡咯、吲哚、哌啶和吡啶等)。重点关注合成方法、催化剂类型、反应路径和反应机理,同时分析了催化剂和氮源对产物分布的影响。该综述为今后生物质基糠醛转化为含氮化合物的研究提供了一些基础信息,为发展更多高效的糠醛催化转化体系提供依据和系统性知识。
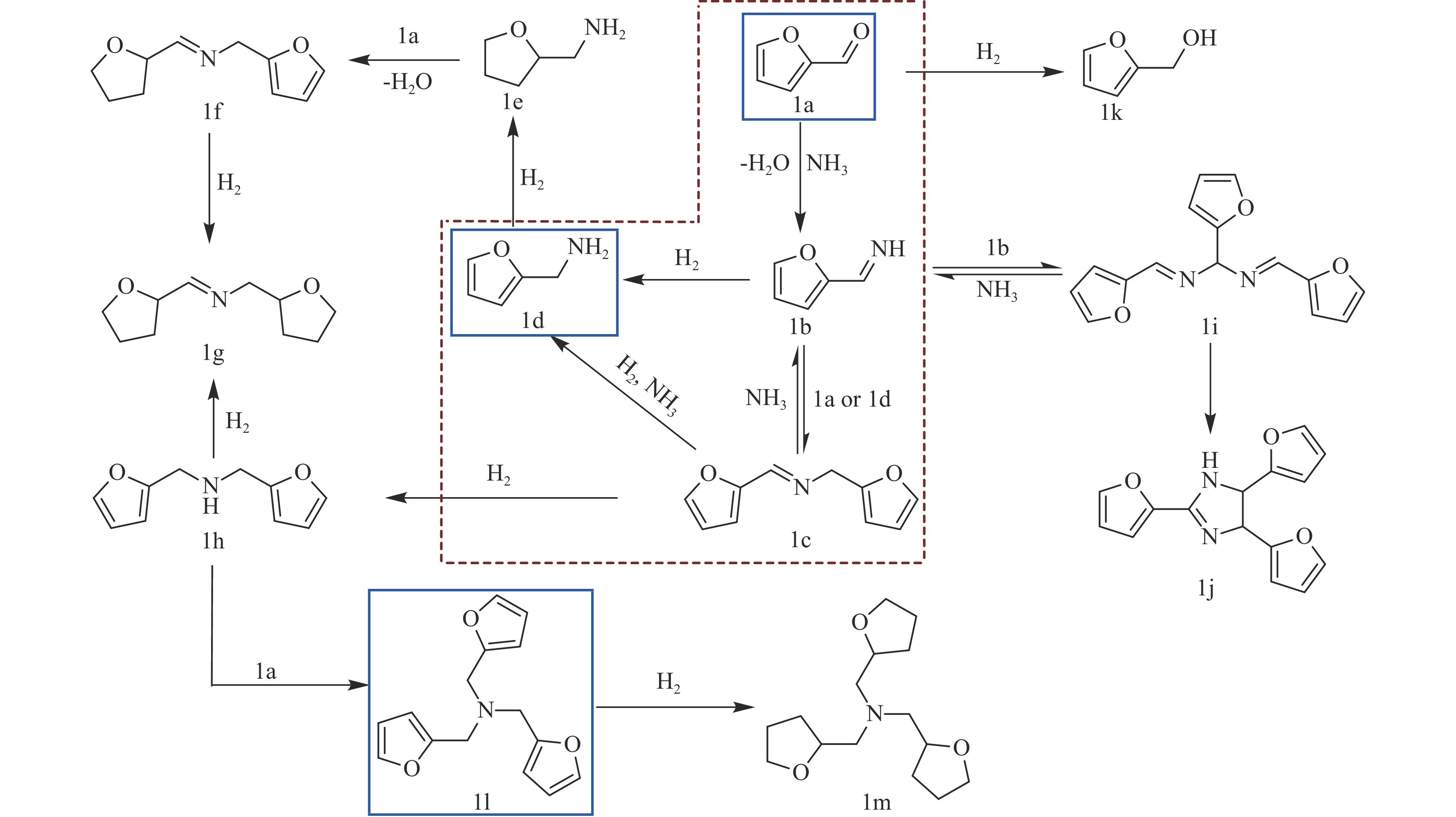
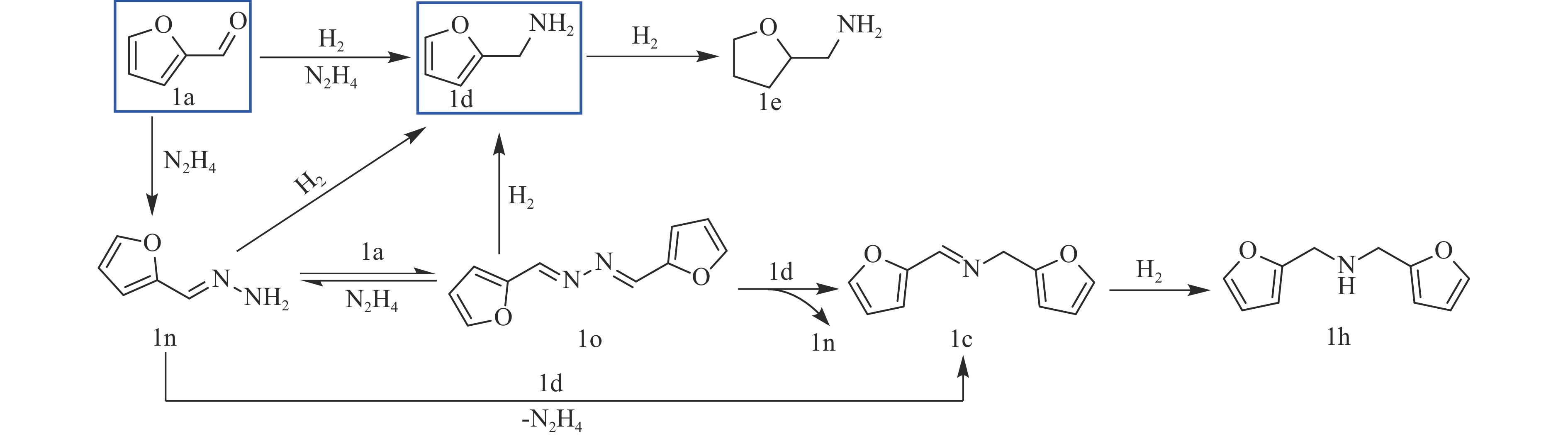
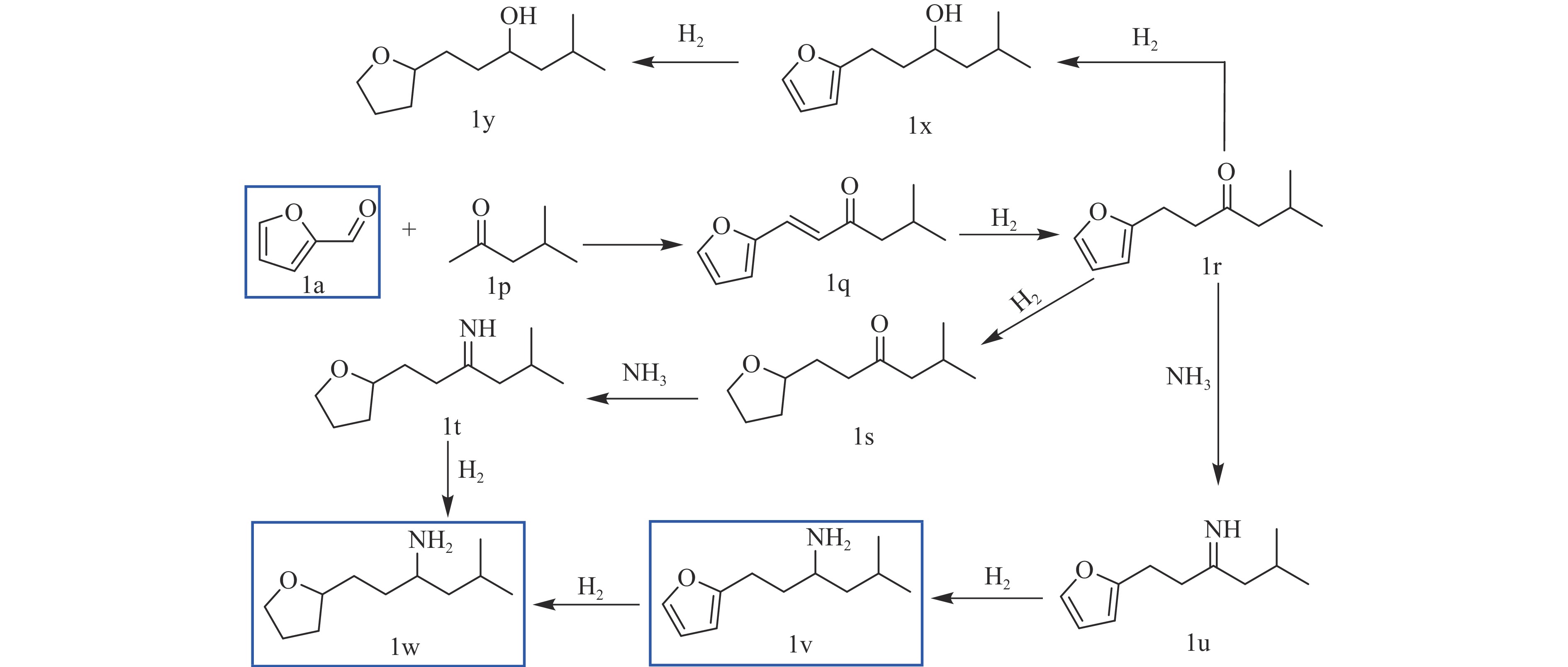

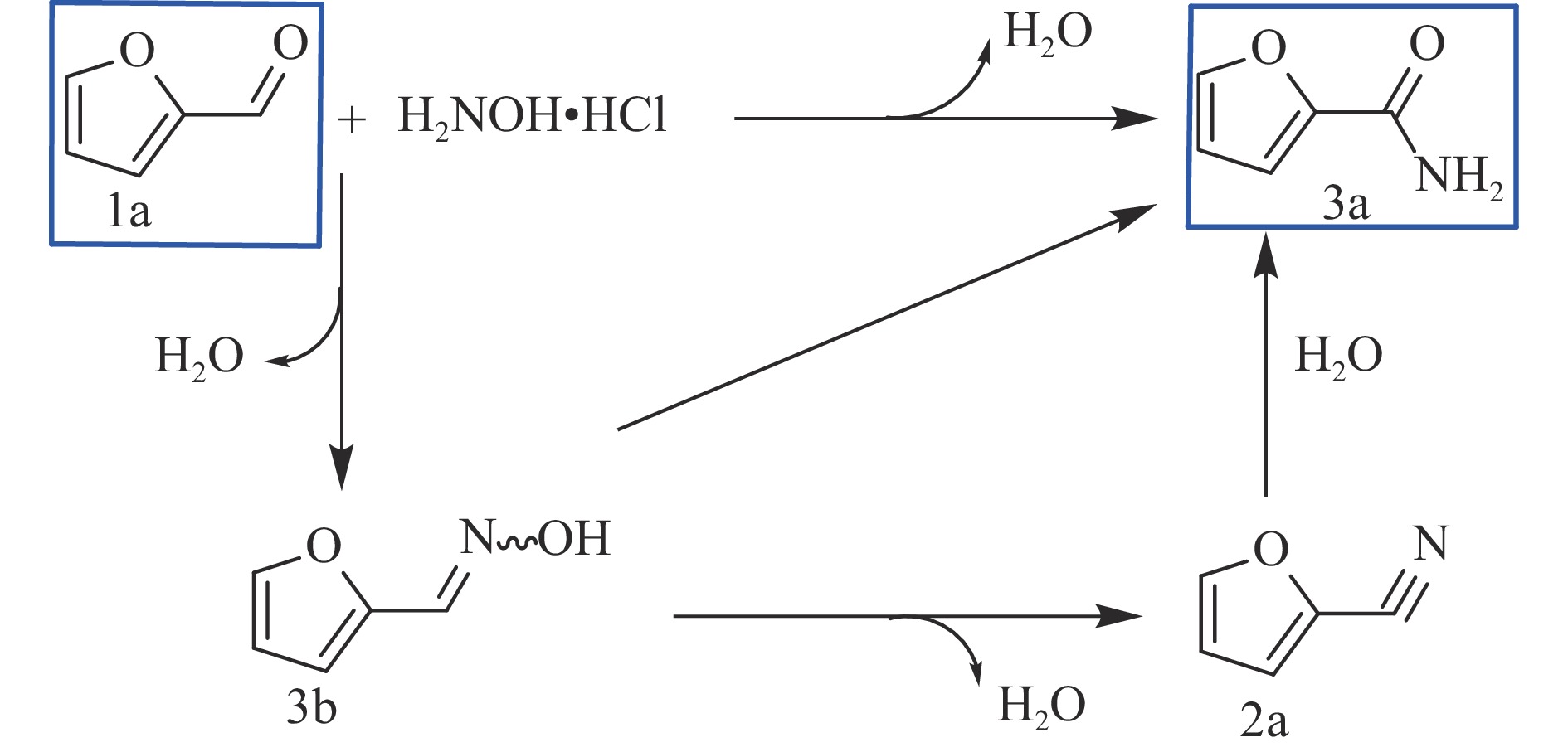
当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60411-6
摘要:
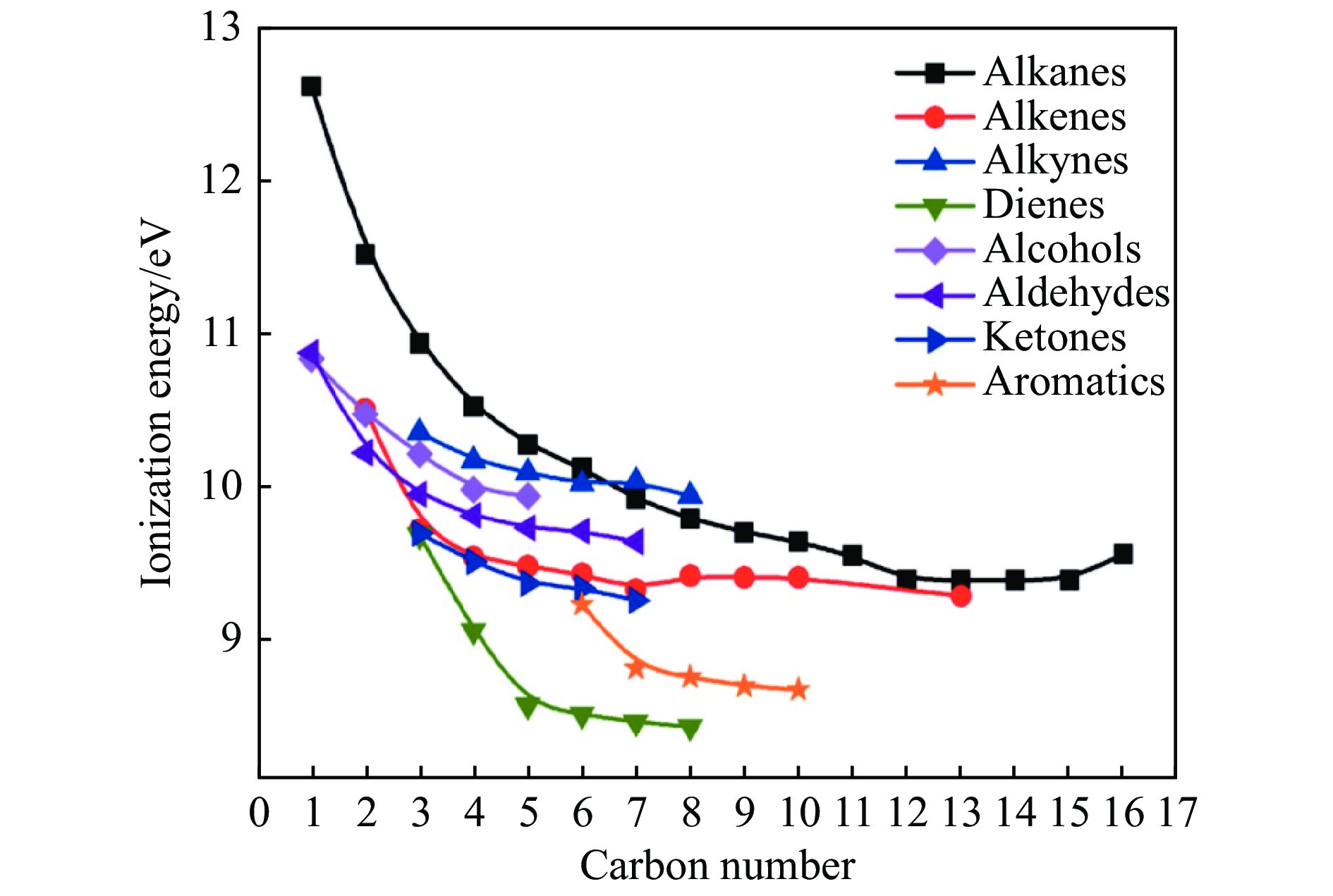
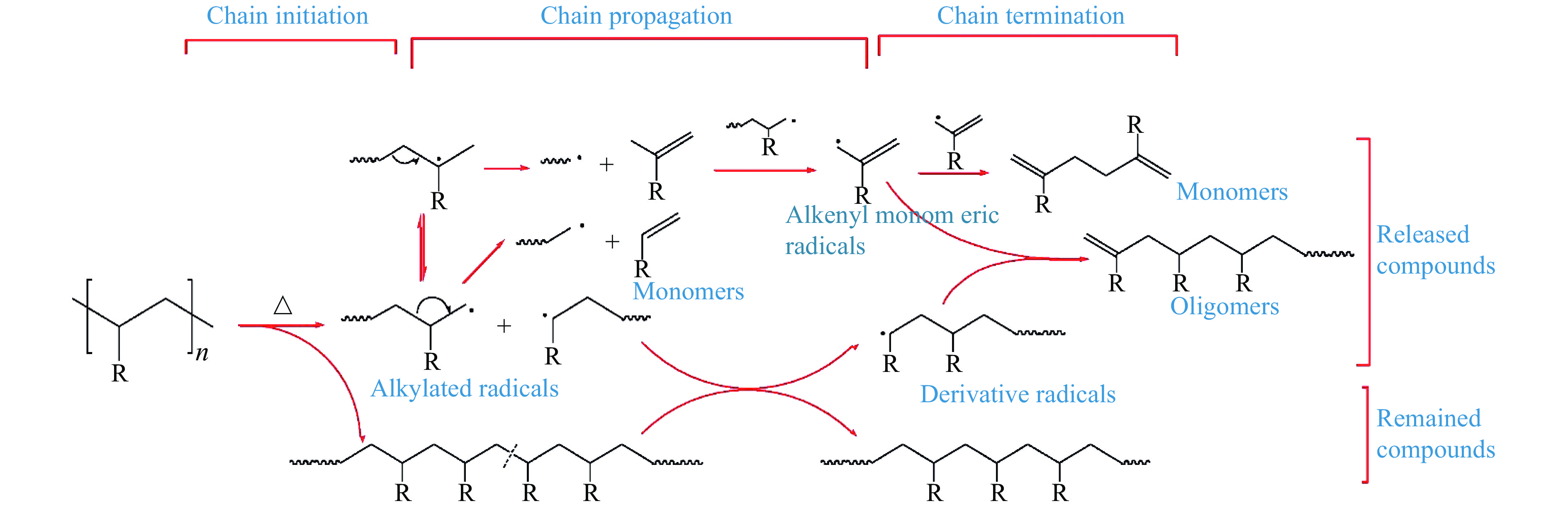
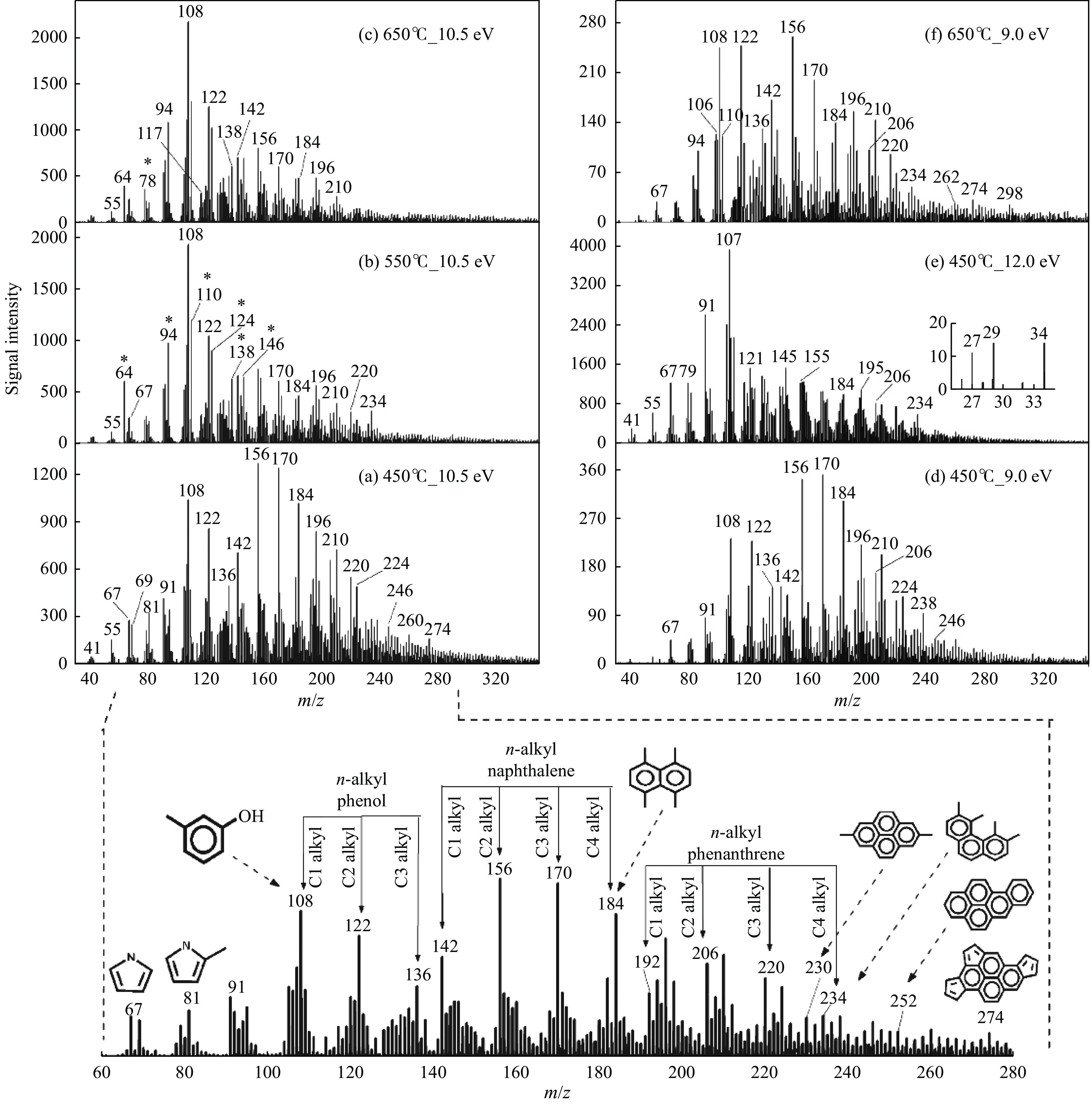
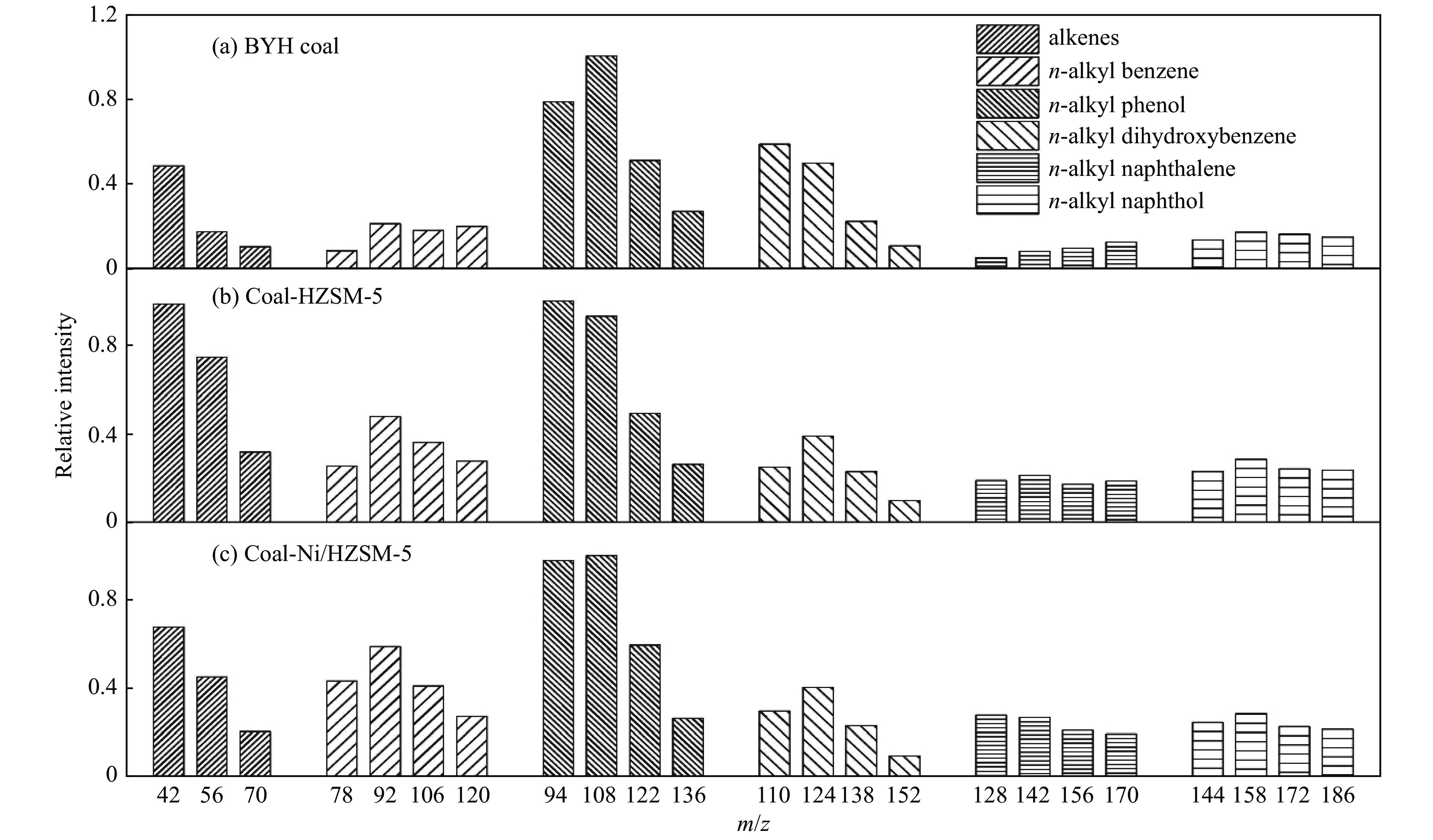
Pyrolysis, an economically viable method, thermochemically converts solid fuel into transportation fuels and value-added chemicals, such as clean gas, liquid fuels, and chemicals, alongside undesirable by-products. Photoionization mass spectrometry (PIMS) is a versatile technique for real-time process analysis, offering 'soft' ionization for complex analytes, detecting and analyzing ions during in-situ pyrolysis. This review focuses on recent applications of PIMS during pyrolysis of solid fuels (i.e. coal, biomass and energetic materials). It summarizes studies on mass spectrometric analysis combined with different reactors and highlights the benefits othrough online PIMS as a diagnostic tool for in situ analysis. It provides an overview of interplay between experimental advancements and models and discusses future perspectives, potential applications in support of mechanistic studies.
Pyrolysis, an economically viable method, thermochemically converts solid fuel into transportation fuels and value-added chemicals, such as clean gas, liquid fuels, and chemicals, alongside undesirable by-products. Photoionization mass spectrometry (PIMS) is a versatile technique for real-time process analysis, offering 'soft' ionization for complex analytes, detecting and analyzing ions during in-situ pyrolysis. This review focuses on recent applications of PIMS during pyrolysis of solid fuels (i.e. coal, biomass and energetic materials). It summarizes studies on mass spectrometric analysis combined with different reactors and highlights the benefits othrough online PIMS as a diagnostic tool for in situ analysis. It provides an overview of interplay between experimental advancements and models and discusses future perspectives, potential applications in support of mechanistic studies.
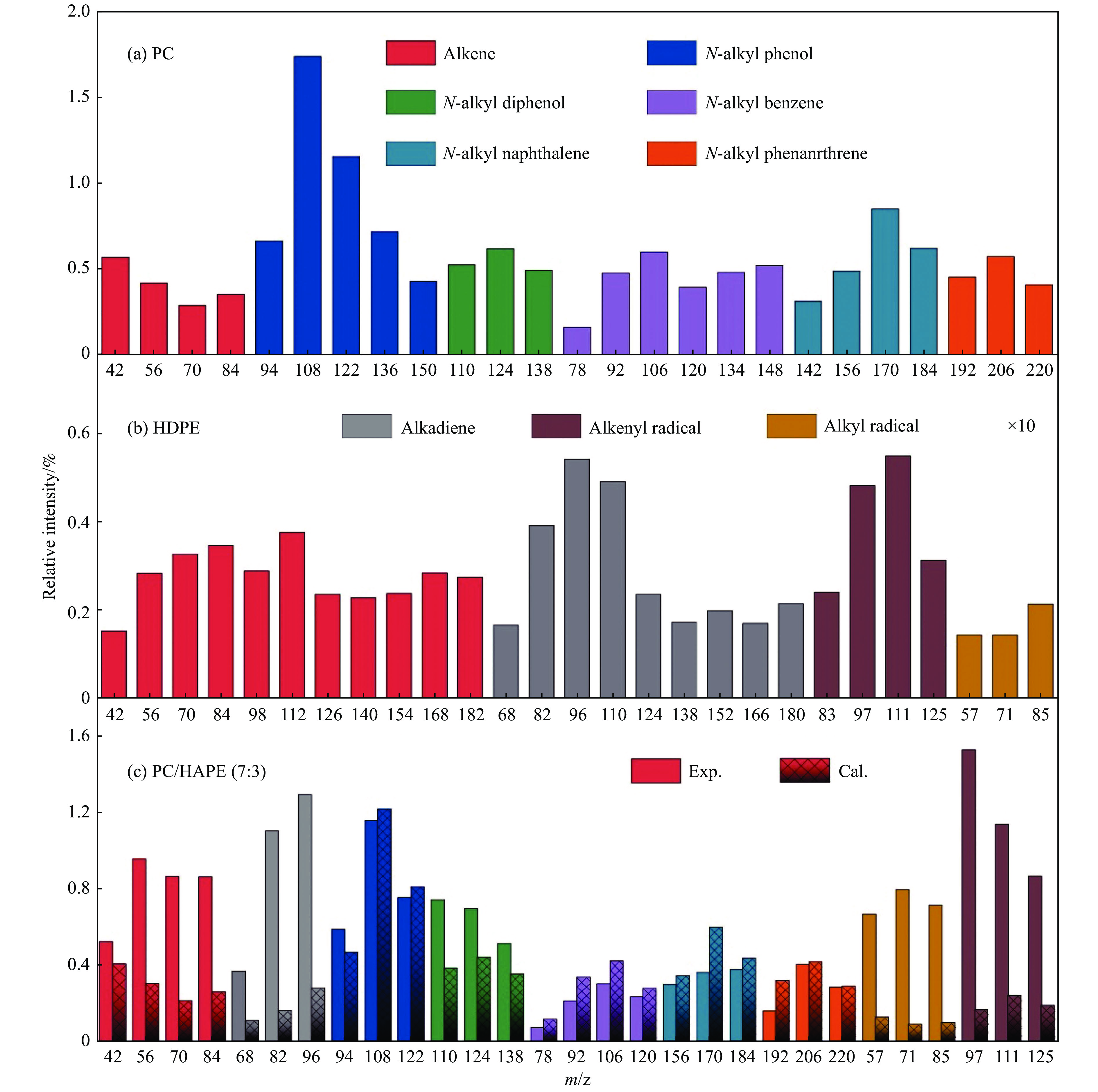
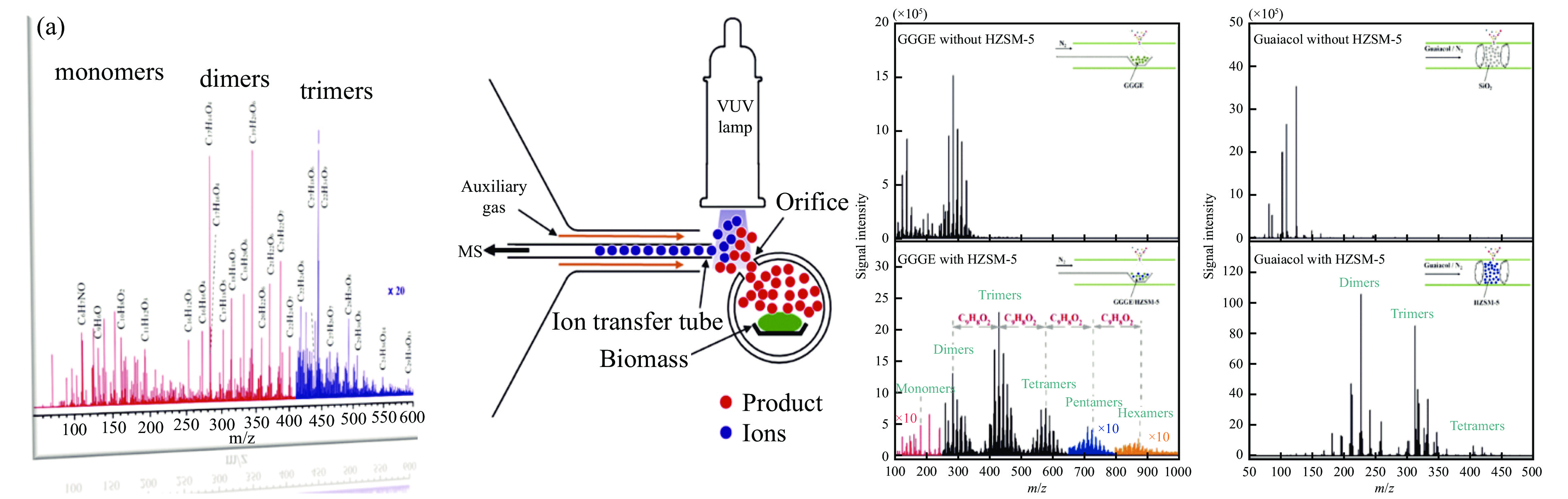
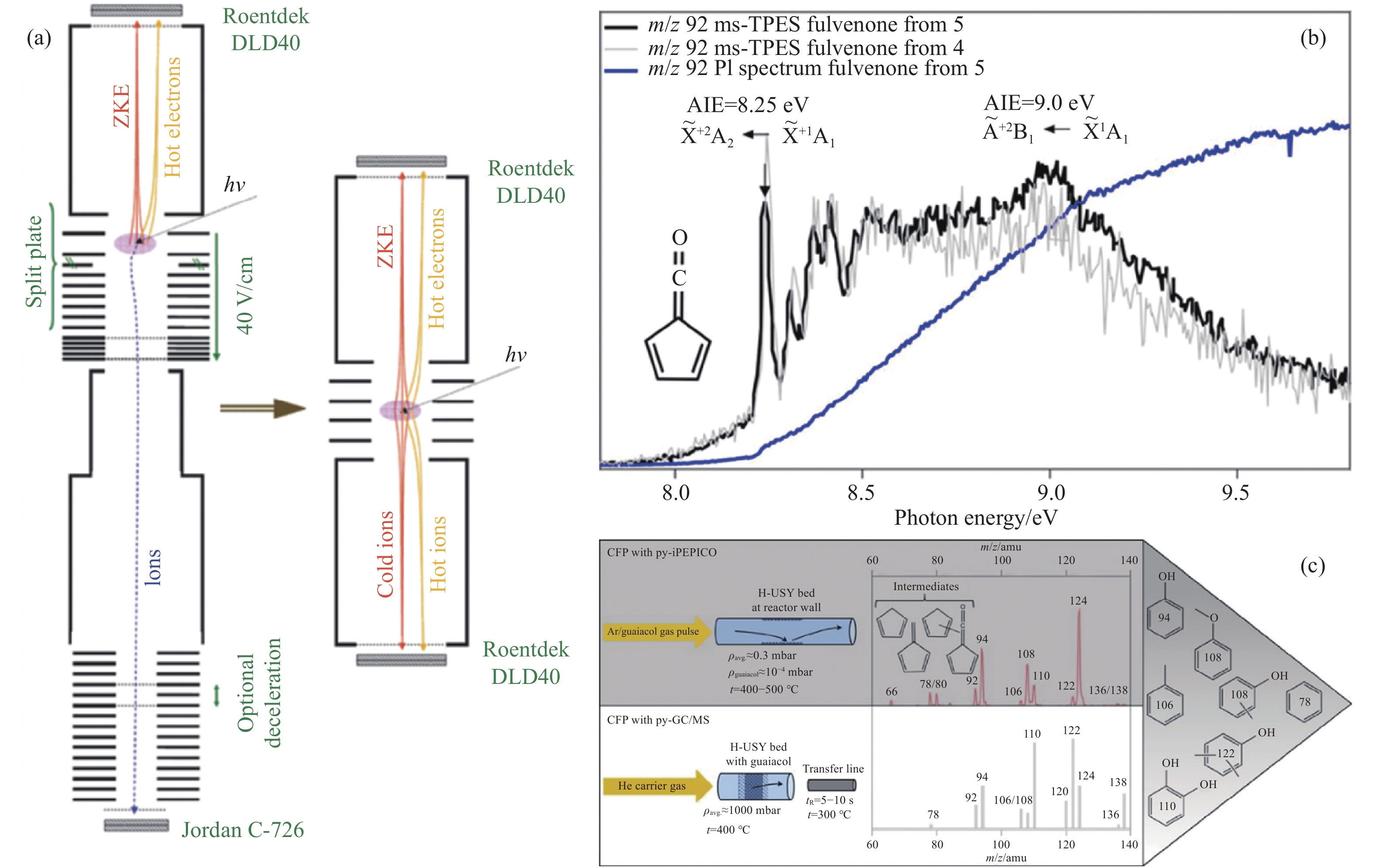

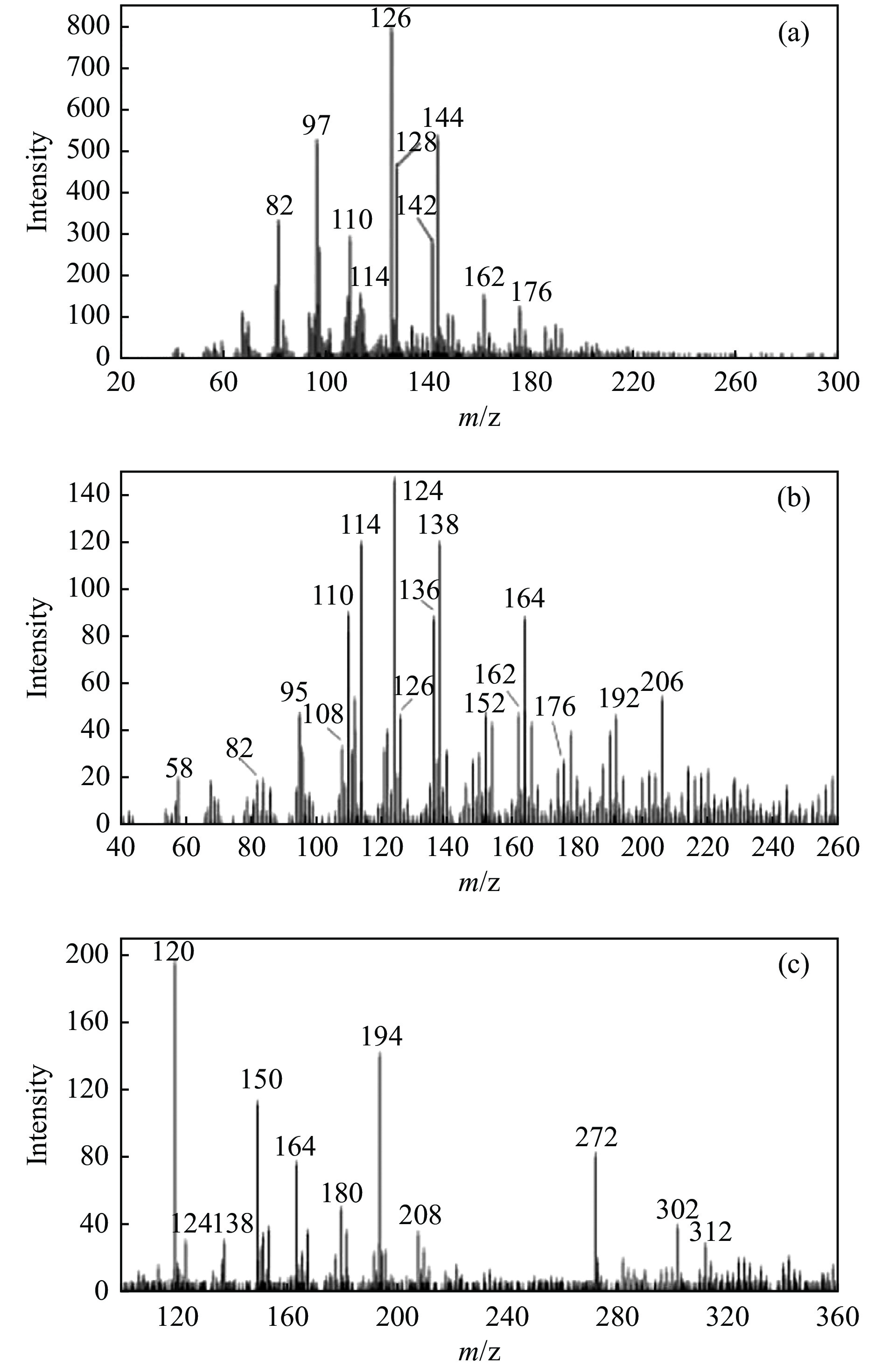

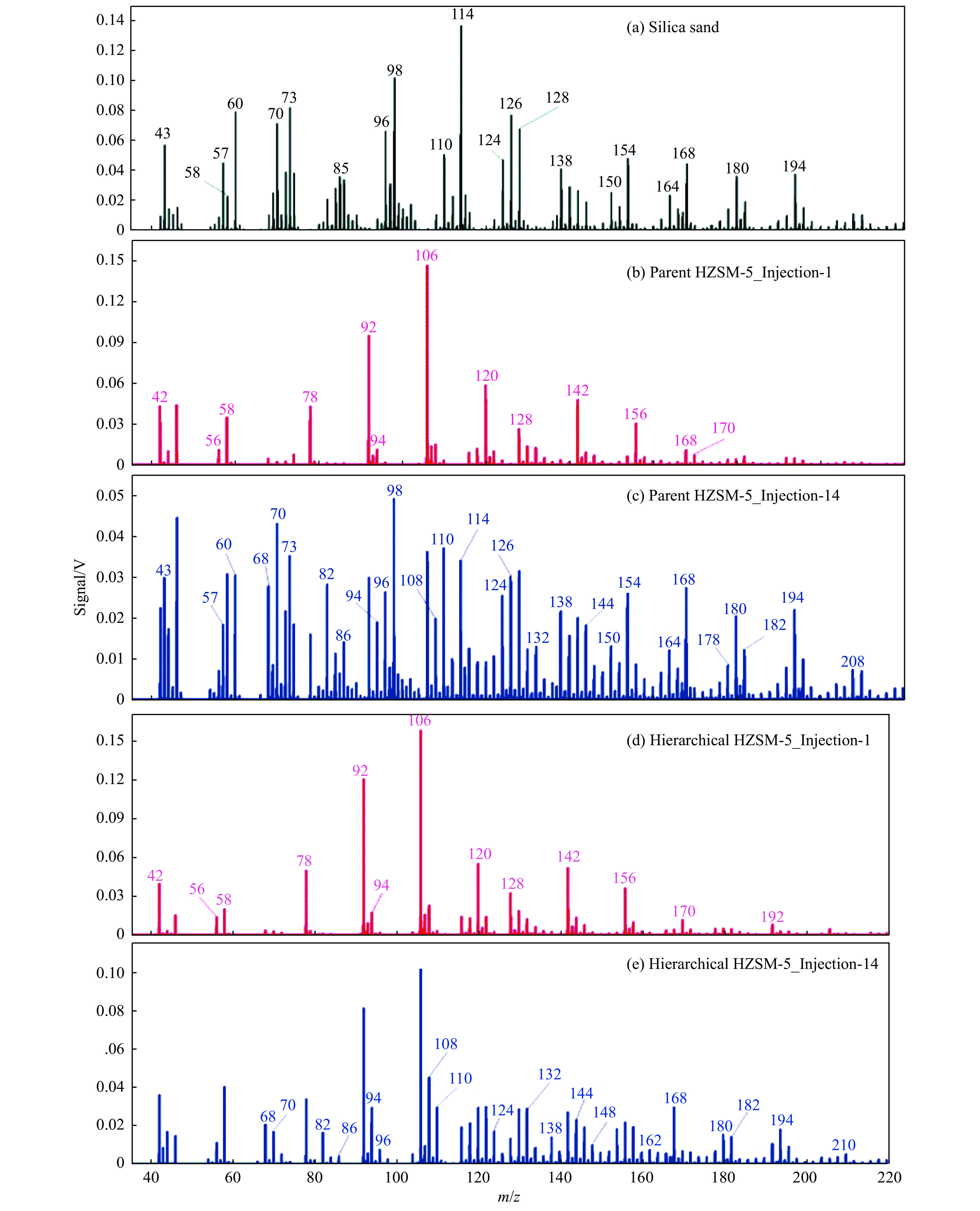
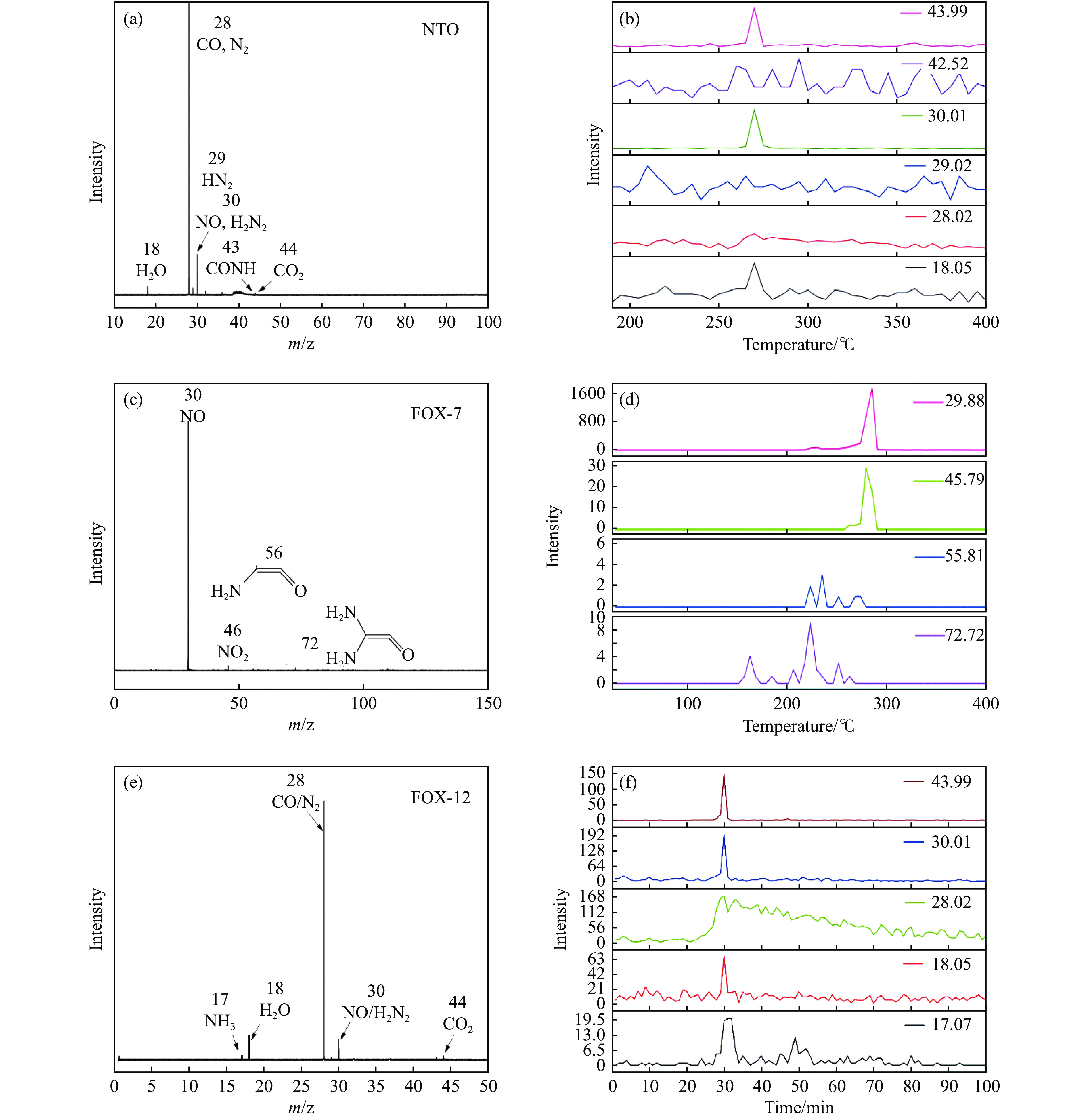
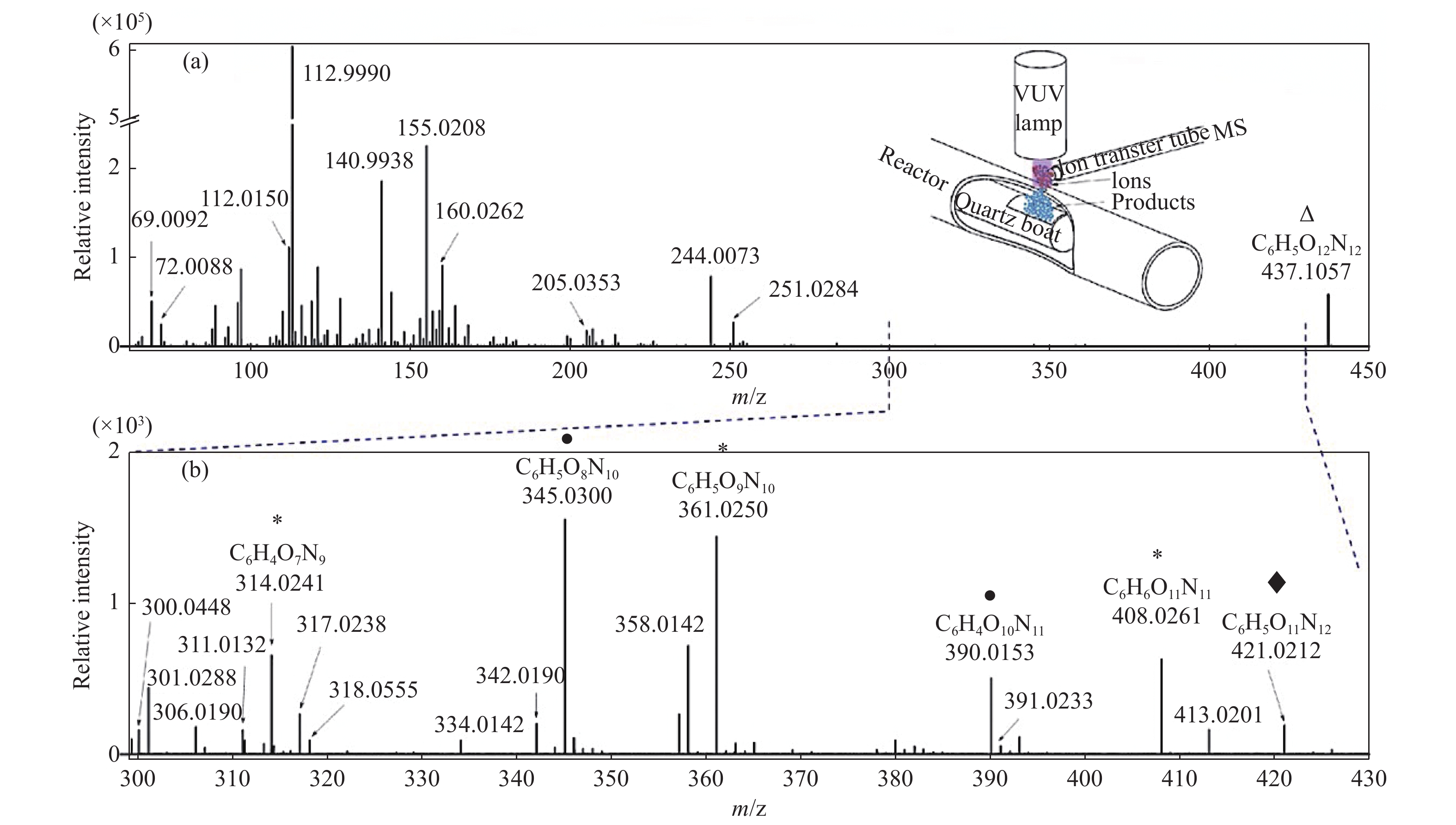
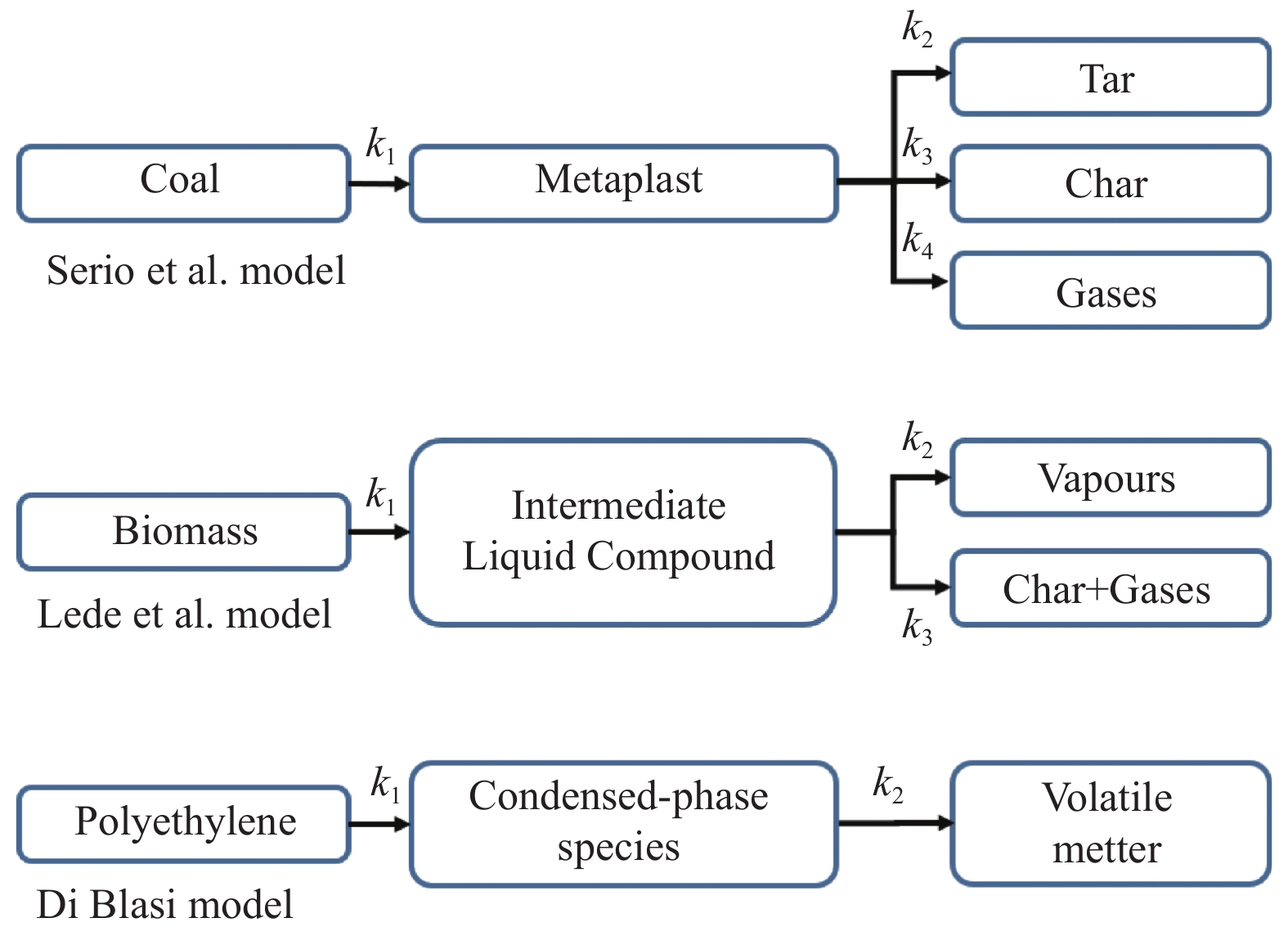

当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60438-X
摘要:
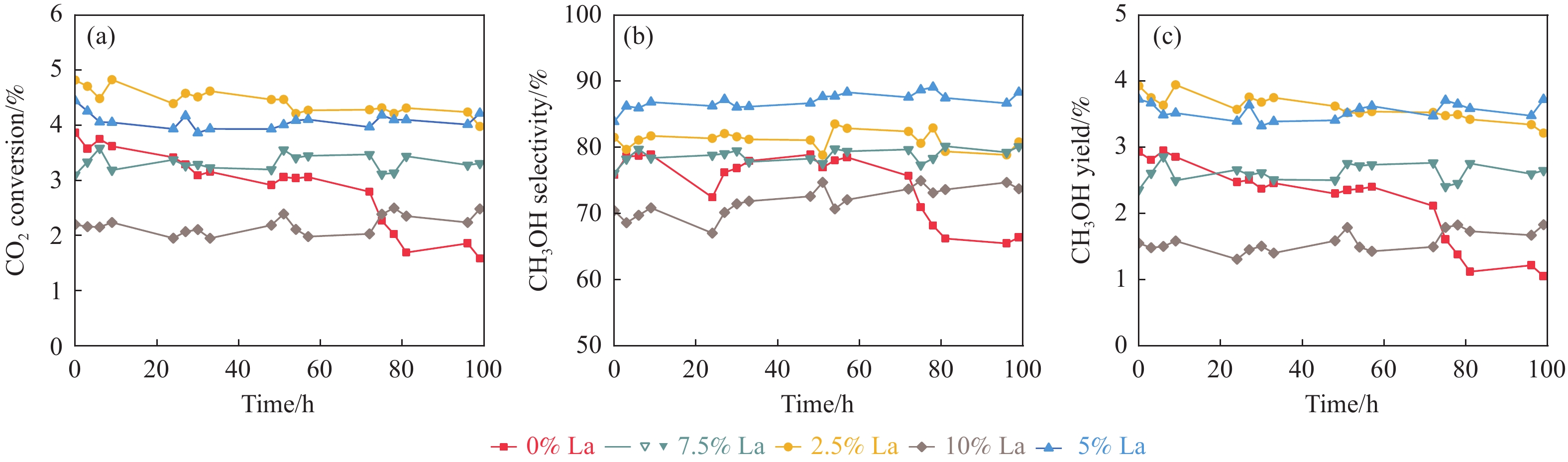
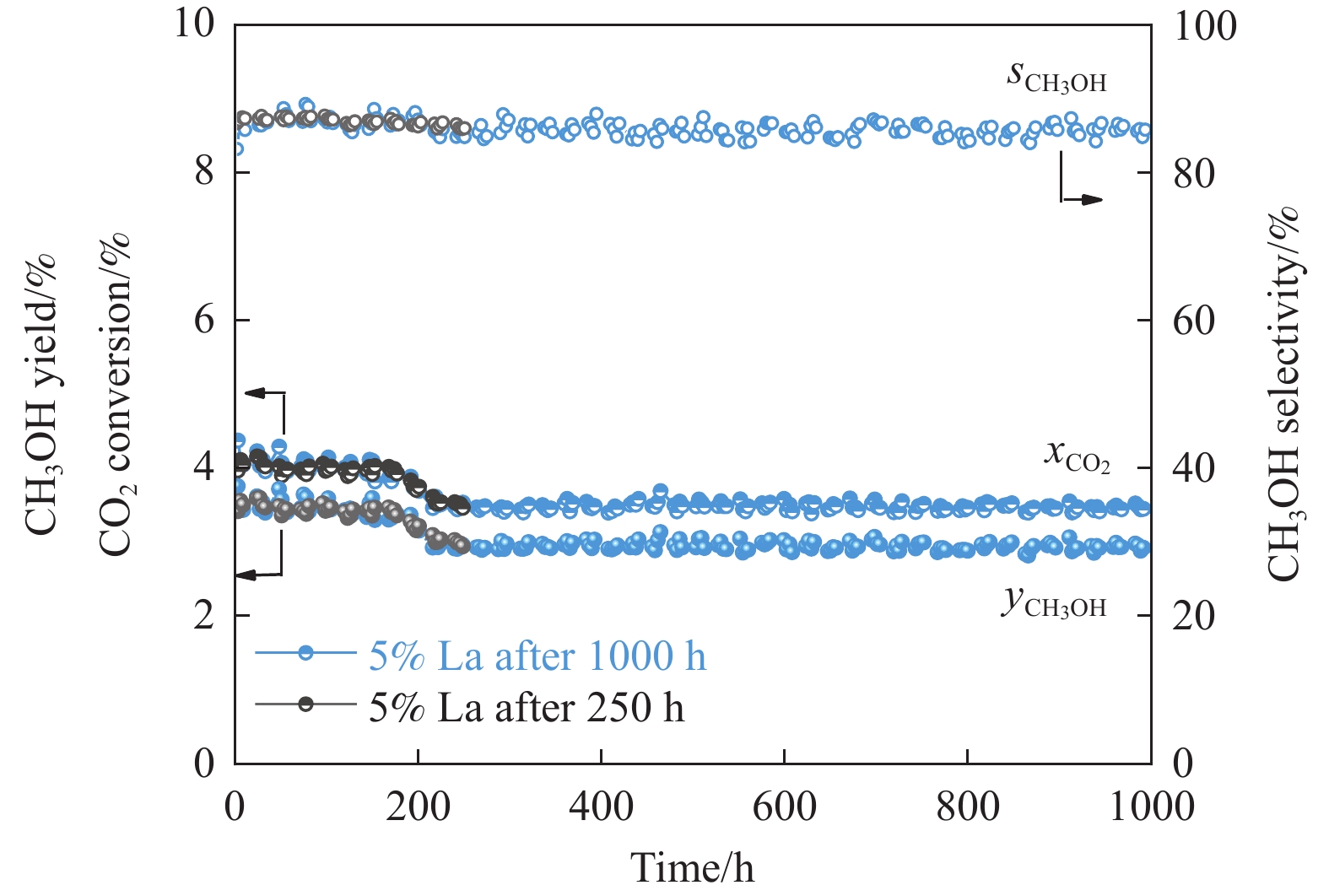
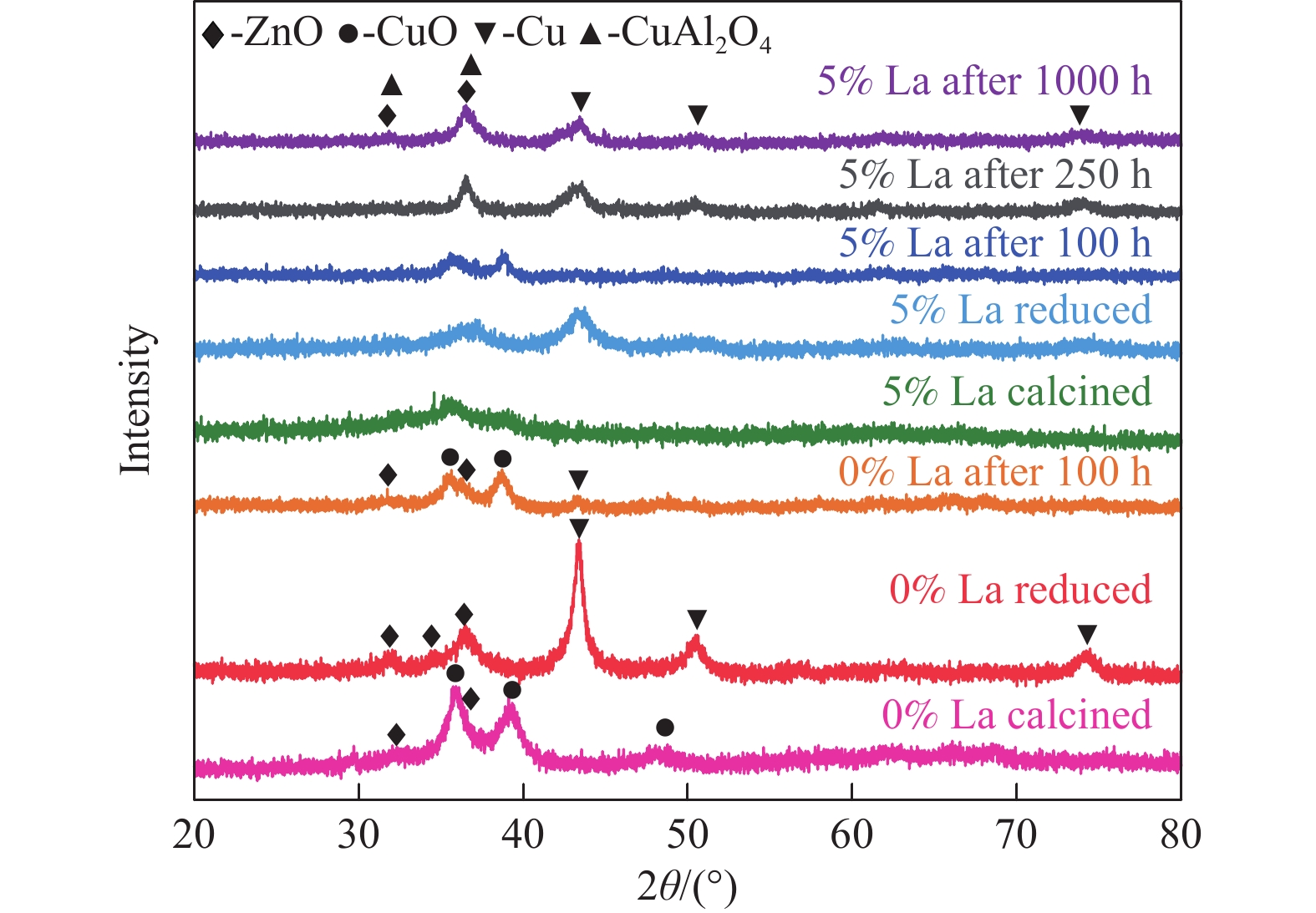
CO2加氢制甲醇反应中Cu/ZnO/Al2O3催化剂的失活是限制其应用的主要原因之一,实验通过向Cu/ZnO/Al2O3催化剂中添加不同含量的La,合成了一系列La改性的Cu/ZnO/Al2O3催化剂,以提高其对 CO2加氢制甲醇反应的催化稳定性。在温度200 ℃,压力3 MPa,空速12000 mL/(g·h)条件下进行的100 h短期稳定性测试中,未改性的Cu/ZnO/Al2O3催化剂在100 h内活性衰减明显,添加La后催化剂稳定性逐渐得到提高,当La添加量为5% 时活性最佳(CO2转化率4%,甲醇选择性85%),并且该催化剂在1000 h长期稳定性测试中表现出较高的稳定性(在190−220 h失活 17% 后保持稳定)。通过X射线衍射(XRD)、X射线光电子能谱(XPS)表征发现,加入5% La提高了Cu/ZnO/Al2O3催化剂中Cu、ZnO的分散度,抑制了催化剂中Cu的烧结;同时稳定了Cu0/+,延缓了催化剂中Cu的氧化,从而提高了催化剂的稳定性。
CO2加氢制甲醇反应中Cu/ZnO/Al2O3催化剂的失活是限制其应用的主要原因之一,实验通过向Cu/ZnO/Al2O3催化剂中添加不同含量的La,合成了一系列La改性的Cu/ZnO/Al2O3催化剂,以提高其对 CO2加氢制甲醇反应的催化稳定性。在温度200 ℃,压力3 MPa,空速12000 mL/(g·h)条件下进行的100 h短期稳定性测试中,未改性的Cu/ZnO/Al2O3催化剂在100 h内活性衰减明显,添加La后催化剂稳定性逐渐得到提高,当La添加量为5% 时活性最佳(CO2转化率4%,甲醇选择性85%),并且该催化剂在1000 h长期稳定性测试中表现出较高的稳定性(在190−220 h失活 17% 后保持稳定)。通过X射线衍射(XRD)、X射线光电子能谱(XPS)表征发现,加入5% La提高了Cu/ZnO/Al2O3催化剂中Cu、ZnO的分散度,抑制了催化剂中Cu的烧结;同时稳定了Cu0/+,延缓了催化剂中Cu的氧化,从而提高了催化剂的稳定性。
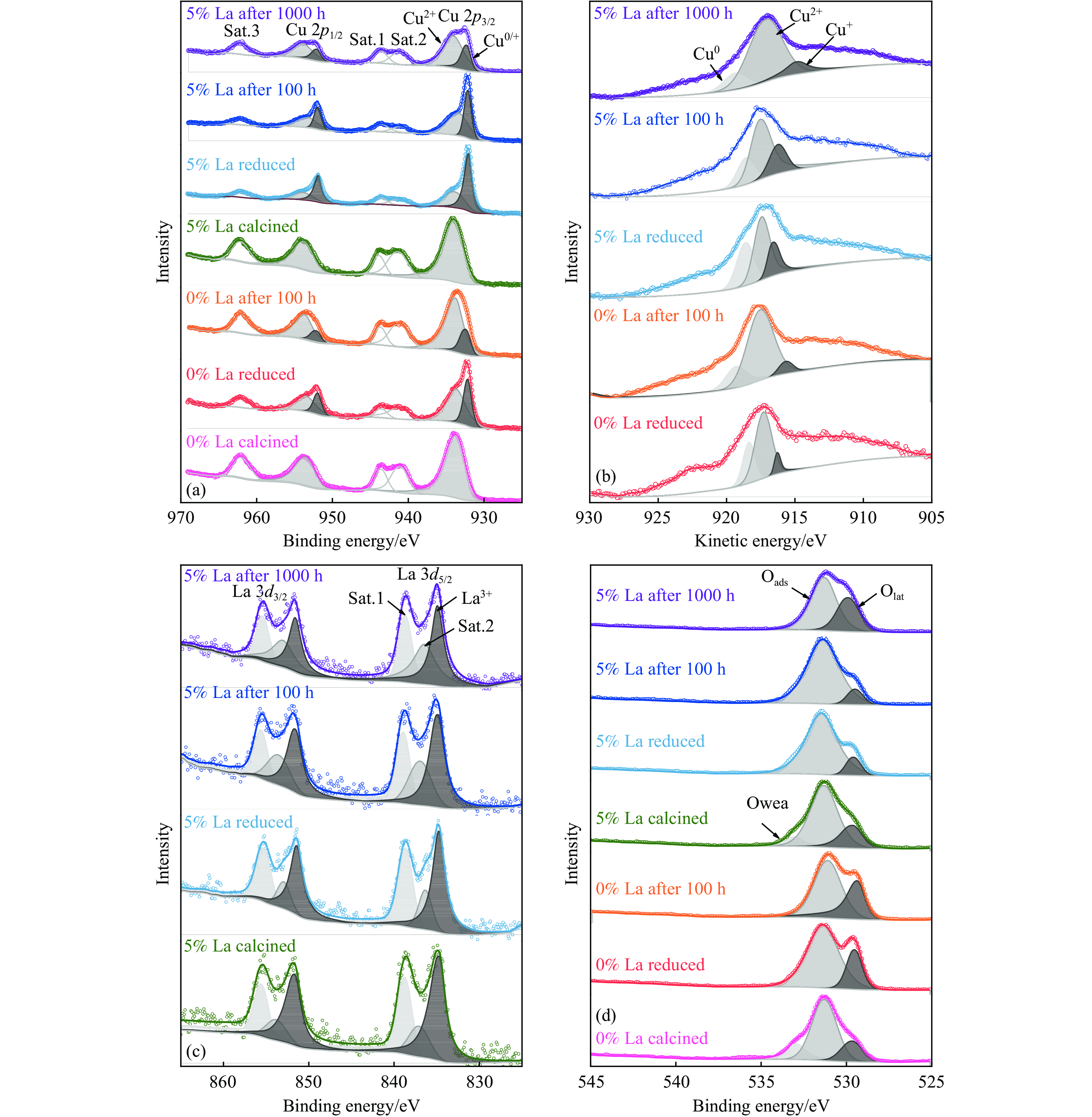
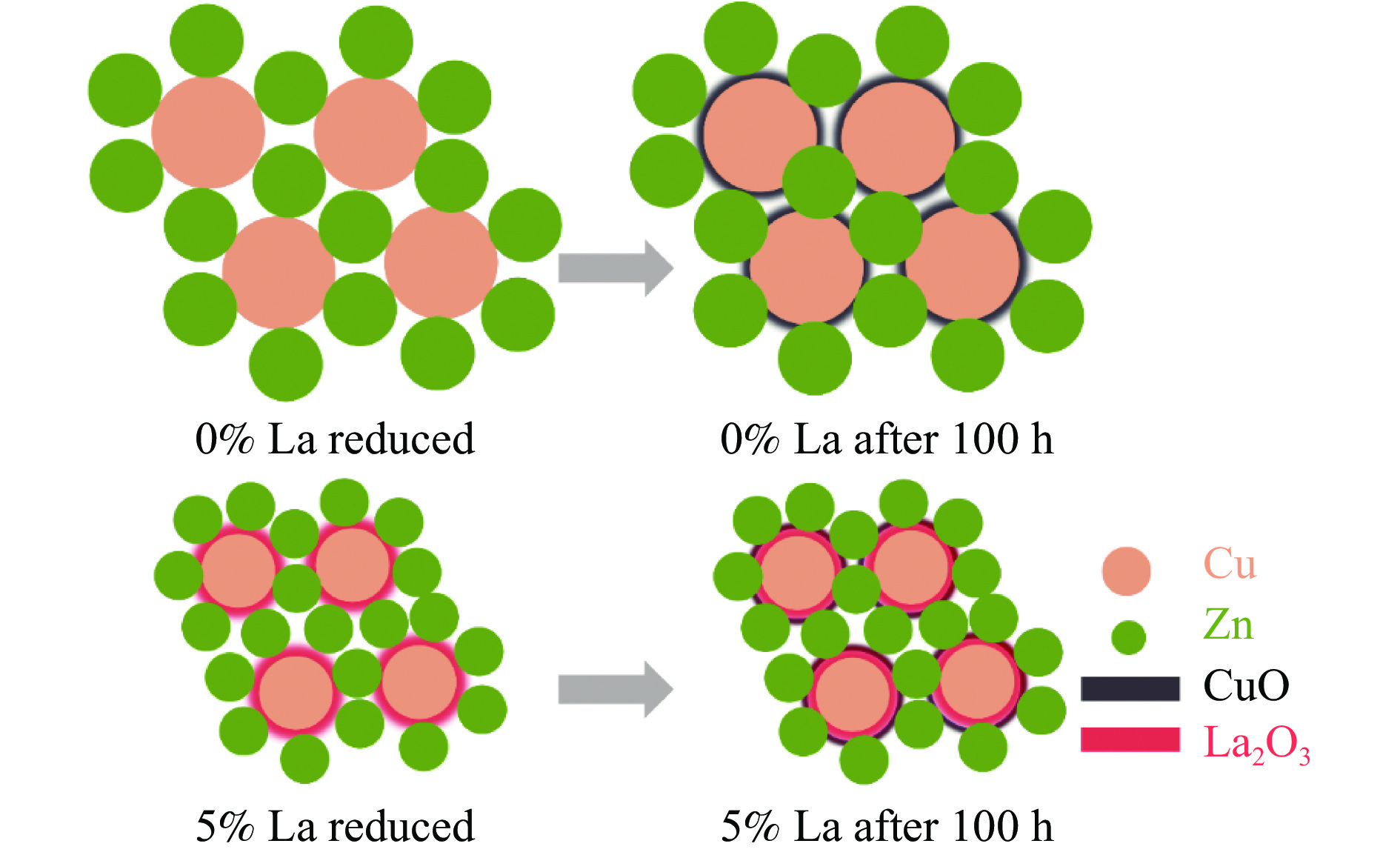
当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024006
摘要:
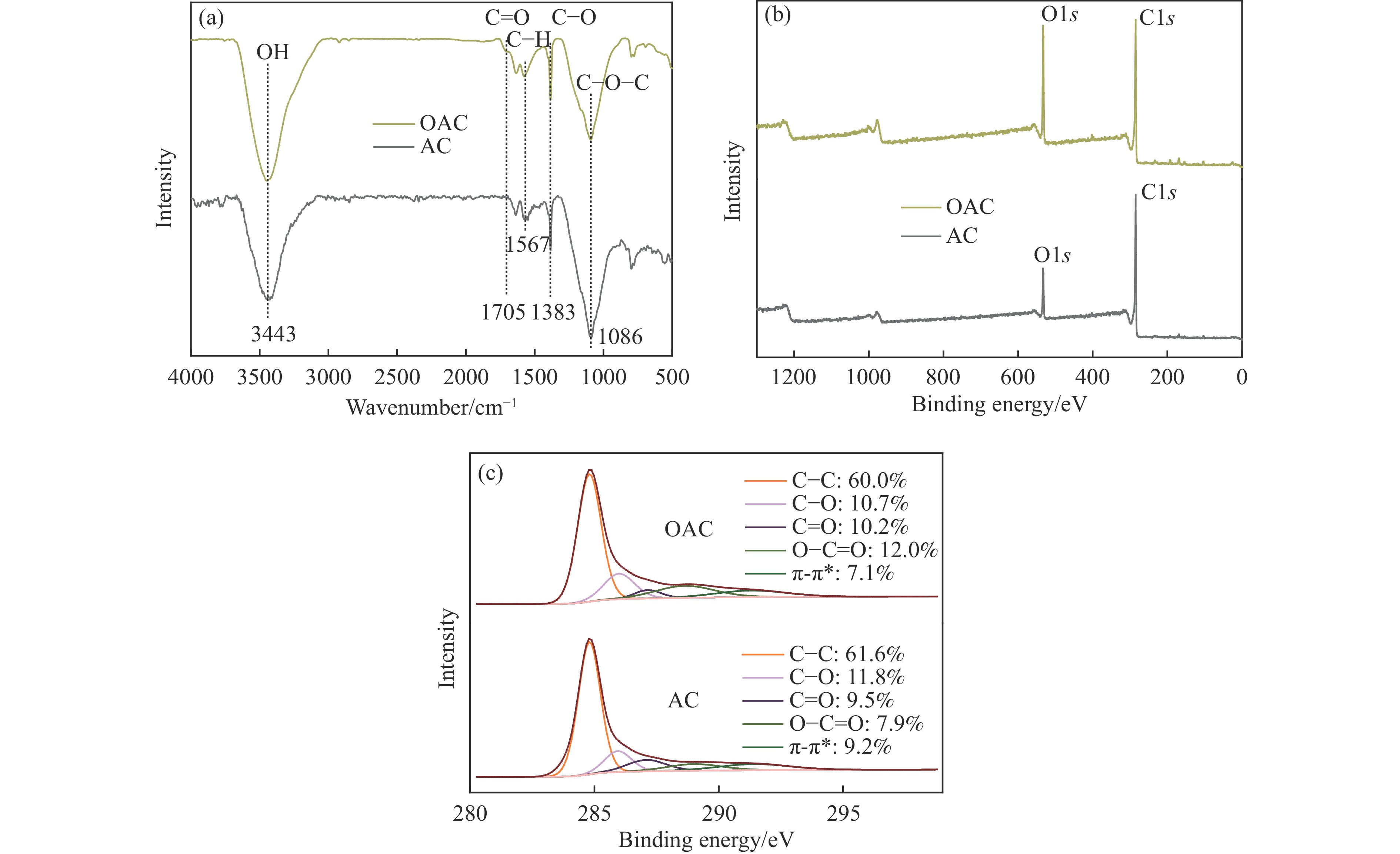
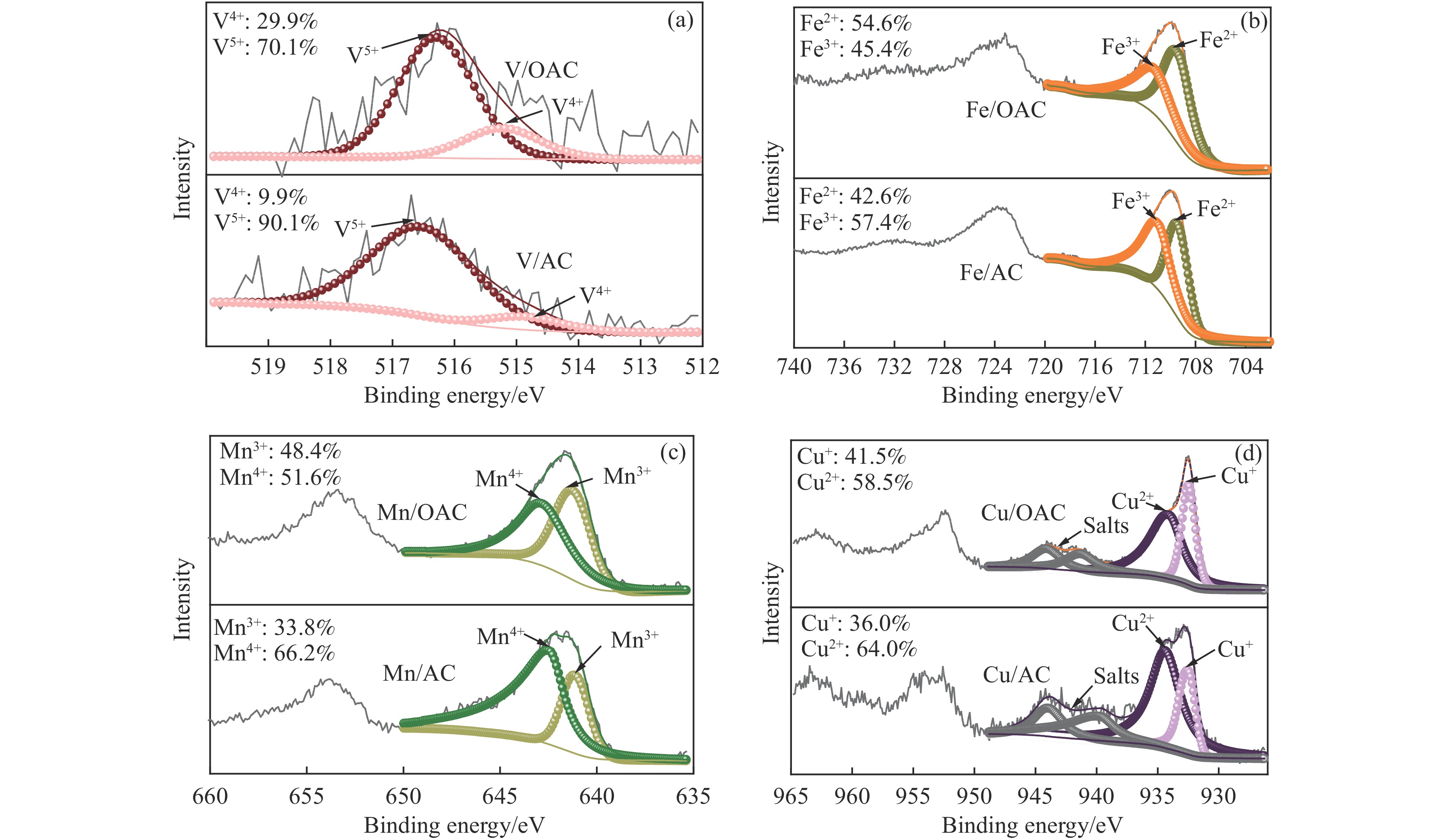
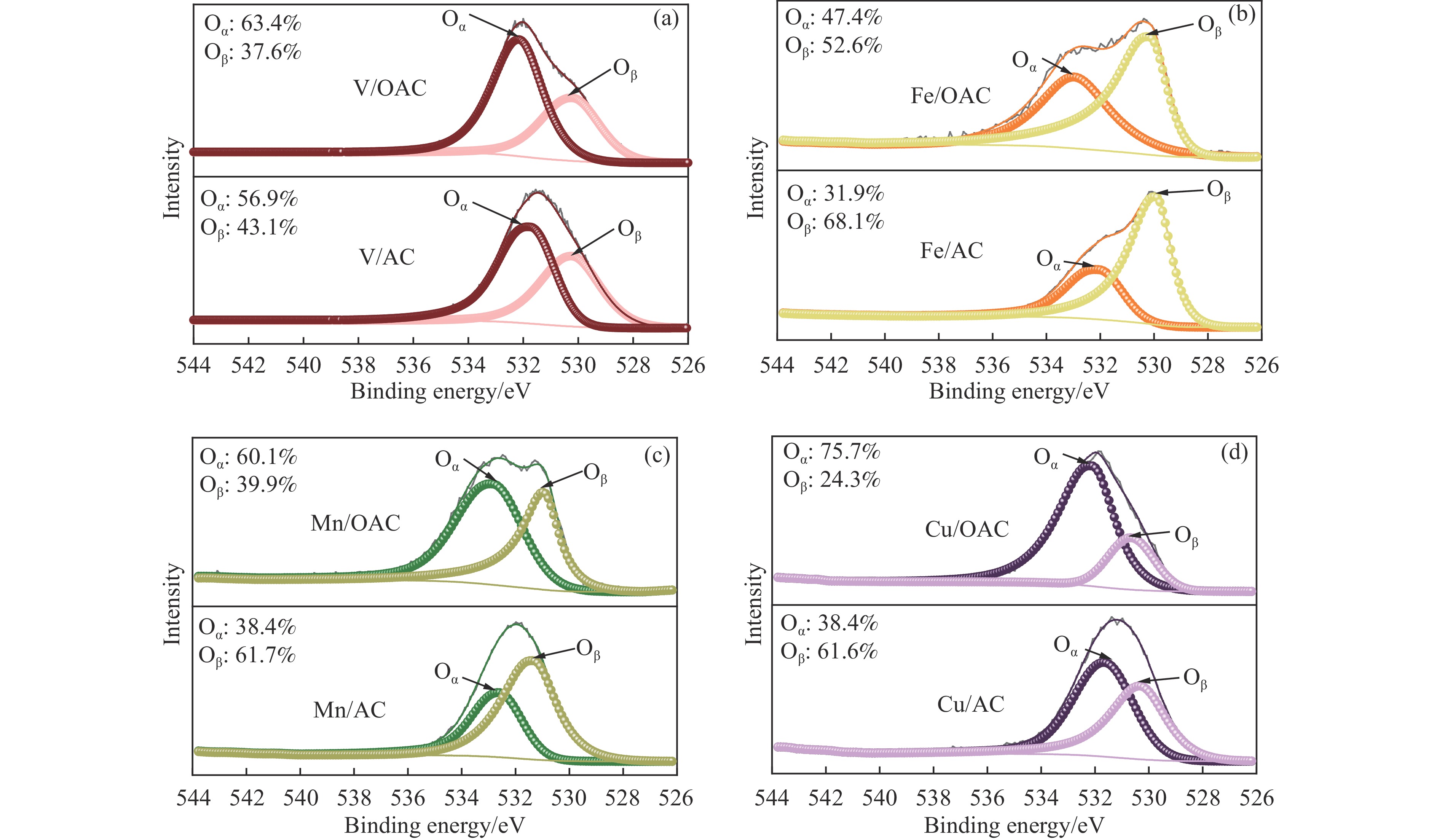
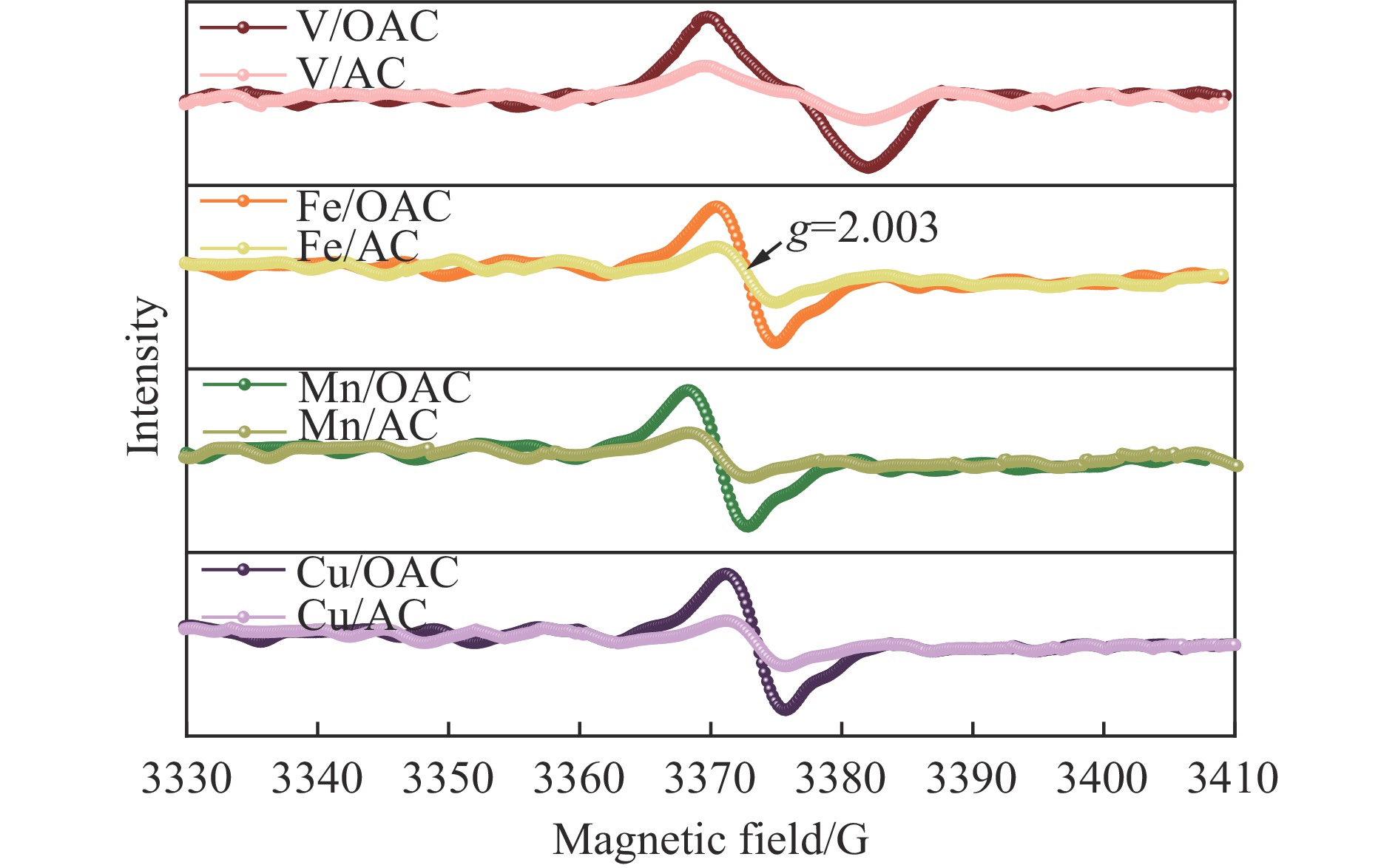
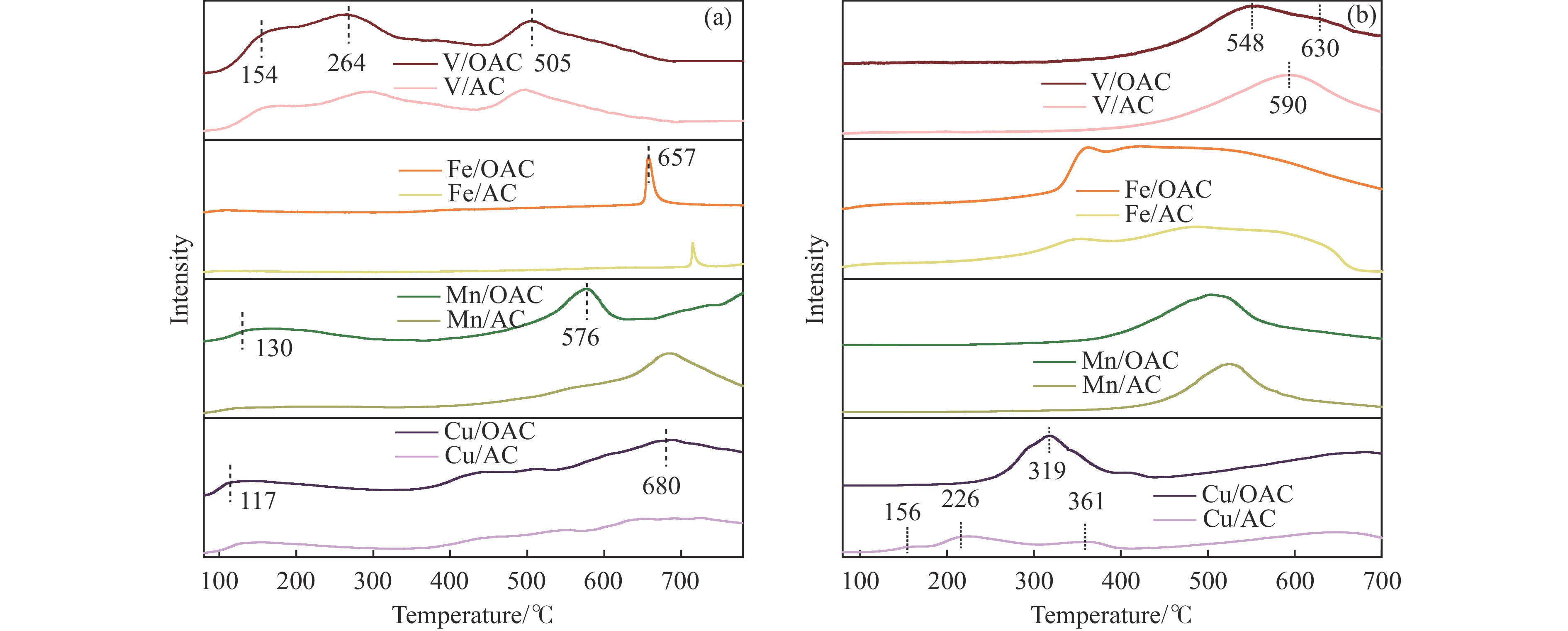
本工作中利用过硫酸铵氧化耦合过渡金属氧化物改性制备V/OAC、Fe/OAC、Mn/OAC、Cu/OAC炭基催化剂,并通过催化活性测试和物理吸附、FT-IR、XPS、NH3-TPD、H2-TPR、EPR等表征手段探究改性炭基催化剂的低温SCR性能增强机制。结果表明,过硫酸铵氧化可向活性炭载体表面引入大量酸性含氧官能团,促进过渡金属氧化物中的氧空位形成,从而提升炭基催化剂的表面酸度和氧化还原性能,进而提升了炭基催化剂低温NH3-SCR性能。本工作发现,过硫酸铵氧化可诱导过渡金属元素(V、Fe、Mn、Cu)的低价态形成。因此,过硫酸铵氧化改性后,活性组分中低价态金属利于NH3-SCR反应的V/OAC、Fe/OAC催化剂性能提升显著,VOx/OAC和FeOx/OAC催化剂在100 ℃下的NO转化率分别从18.2%提升到34.8%和从34.2%提升到55.6%;而活性组分中高价态金属有利NH3-SCR反应的Mn/OAC和Cu/OAC催化剂性能提升有限,100 ℃下的NO转化率仅从61.4%提升到70.4%和61.3%提升到69.7%。本工作总结了过硫酸铵氧化改性对炭基催化剂表面金属价态的调控作用,有助于深入认识过硫酸铵氧化改性对炭基催化剂物化性质的调控规律,为高效炭基脱硝催化剂的开发提供指导和参考。
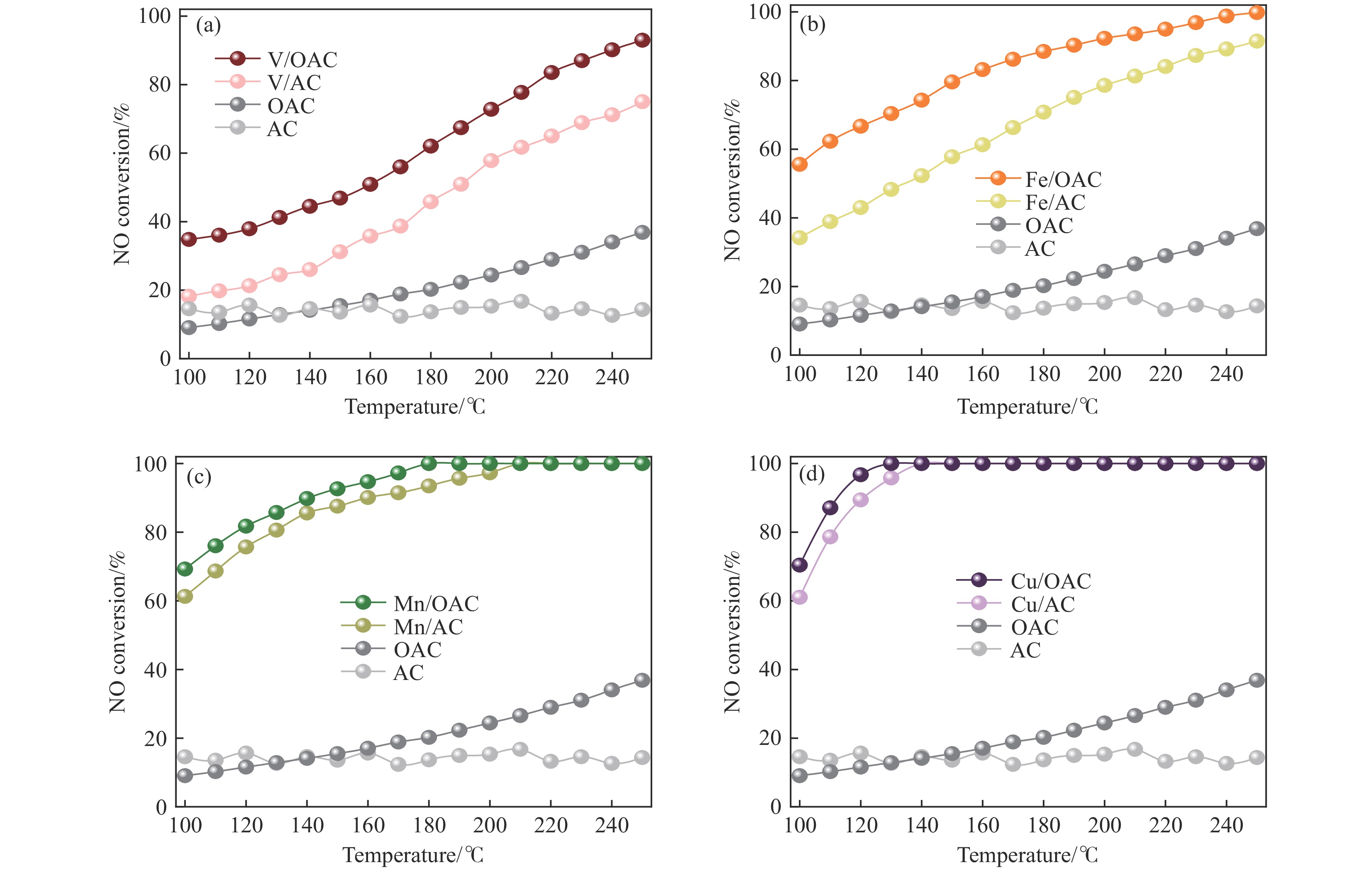
本工作中利用过硫酸铵氧化耦合过渡金属氧化物改性制备V/OAC、Fe/OAC、Mn/OAC、Cu/OAC炭基催化剂,并通过催化活性测试和物理吸附、FT-IR、XPS、NH3-TPD、H2-TPR、EPR等表征手段探究改性炭基催化剂的低温SCR性能增强机制。结果表明,过硫酸铵氧化可向活性炭载体表面引入大量酸性含氧官能团,促进过渡金属氧化物中的氧空位形成,从而提升炭基催化剂的表面酸度和氧化还原性能,进而提升了炭基催化剂低温NH3-SCR性能。本工作发现,过硫酸铵氧化可诱导过渡金属元素(V、Fe、Mn、Cu)的低价态形成。因此,过硫酸铵氧化改性后,活性组分中低价态金属利于NH3-SCR反应的V/OAC、Fe/OAC催化剂性能提升显著,VOx/OAC和FeOx/OAC催化剂在100 ℃下的NO转化率分别从18.2%提升到34.8%和从34.2%提升到55.6%;而活性组分中高价态金属有利NH3-SCR反应的Mn/OAC和Cu/OAC催化剂性能提升有限,100 ℃下的NO转化率仅从61.4%提升到70.4%和61.3%提升到69.7%。本工作总结了过硫酸铵氧化改性对炭基催化剂表面金属价态的调控作用,有助于深入认识过硫酸铵氧化改性对炭基催化剂物化性质的调控规律,为高效炭基脱硝催化剂的开发提供指导和参考。
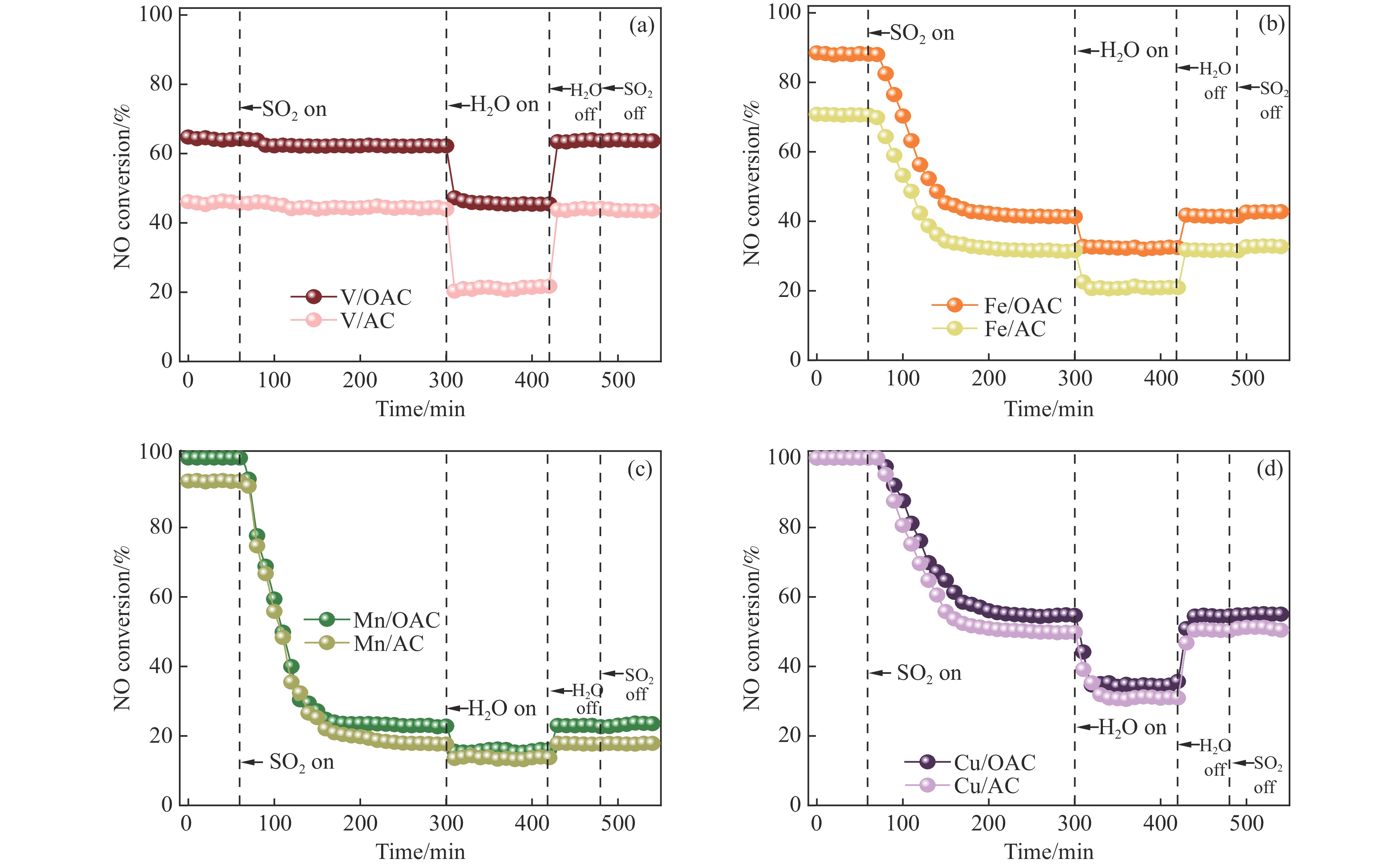

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024008
摘要:
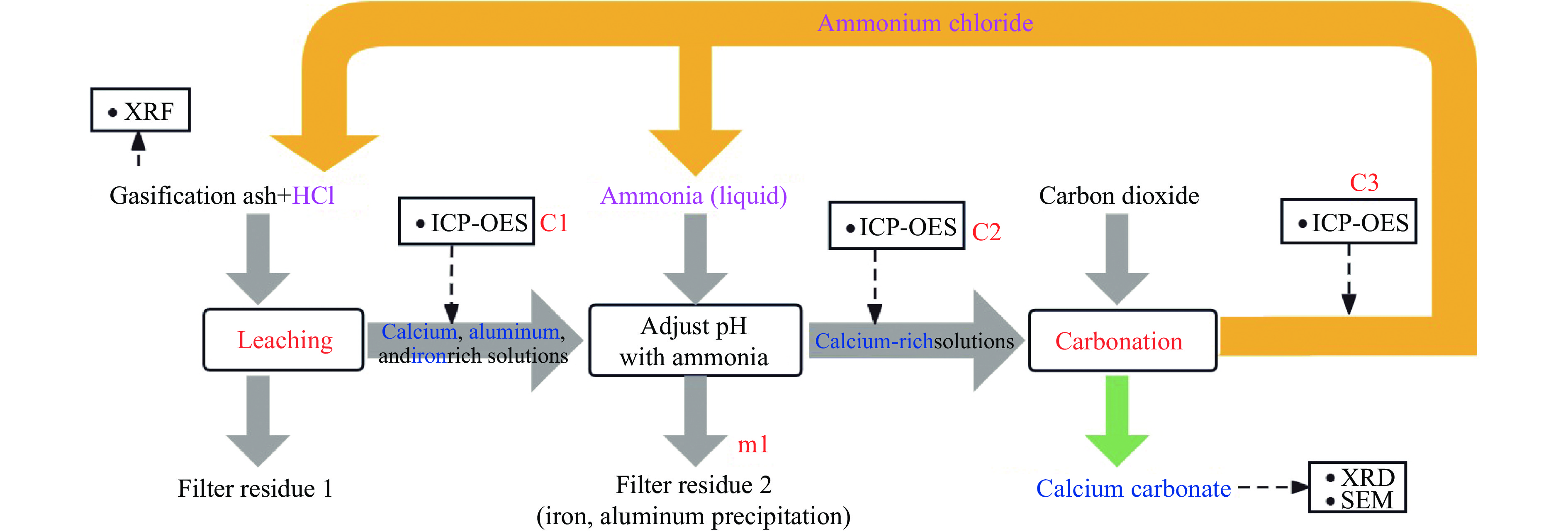
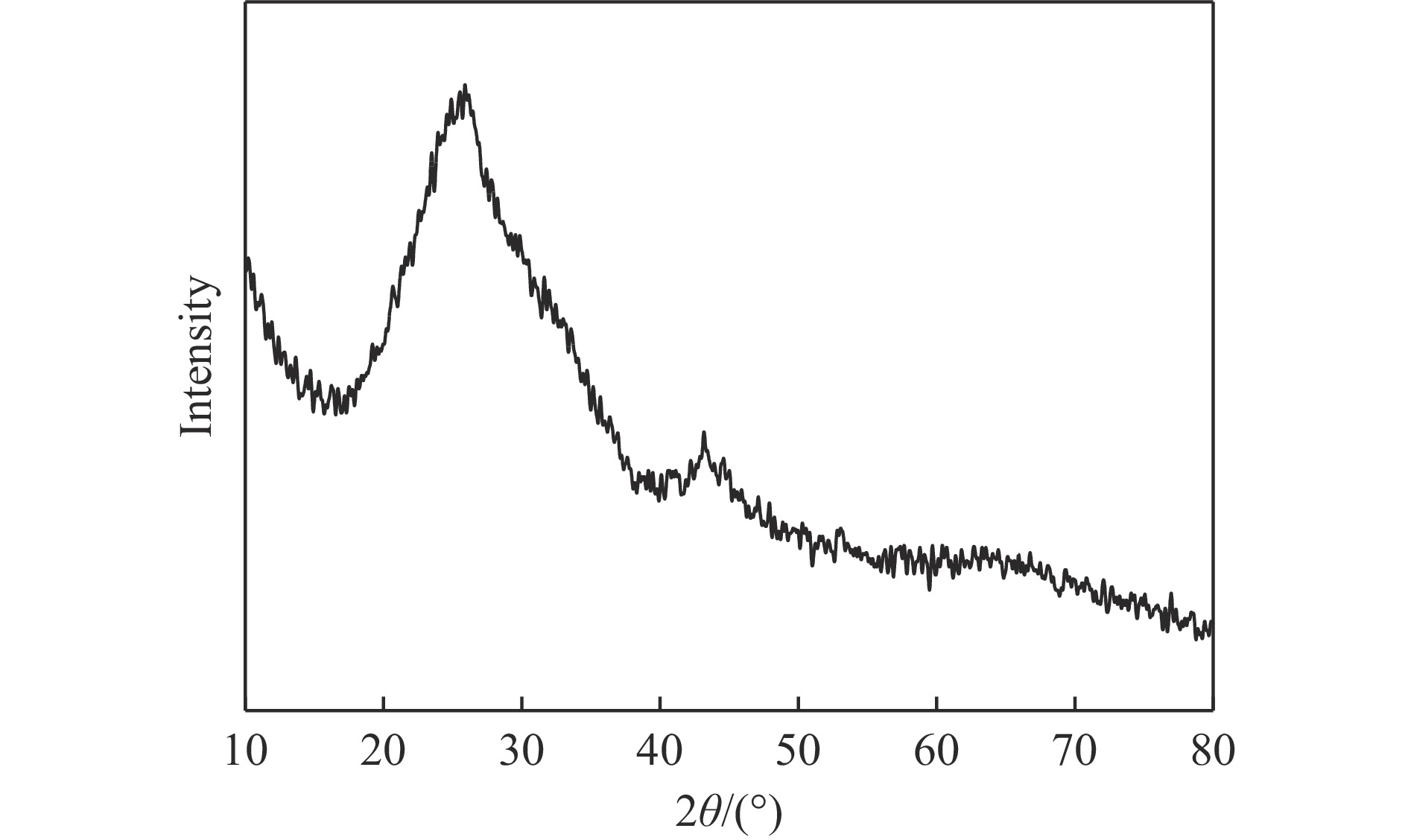
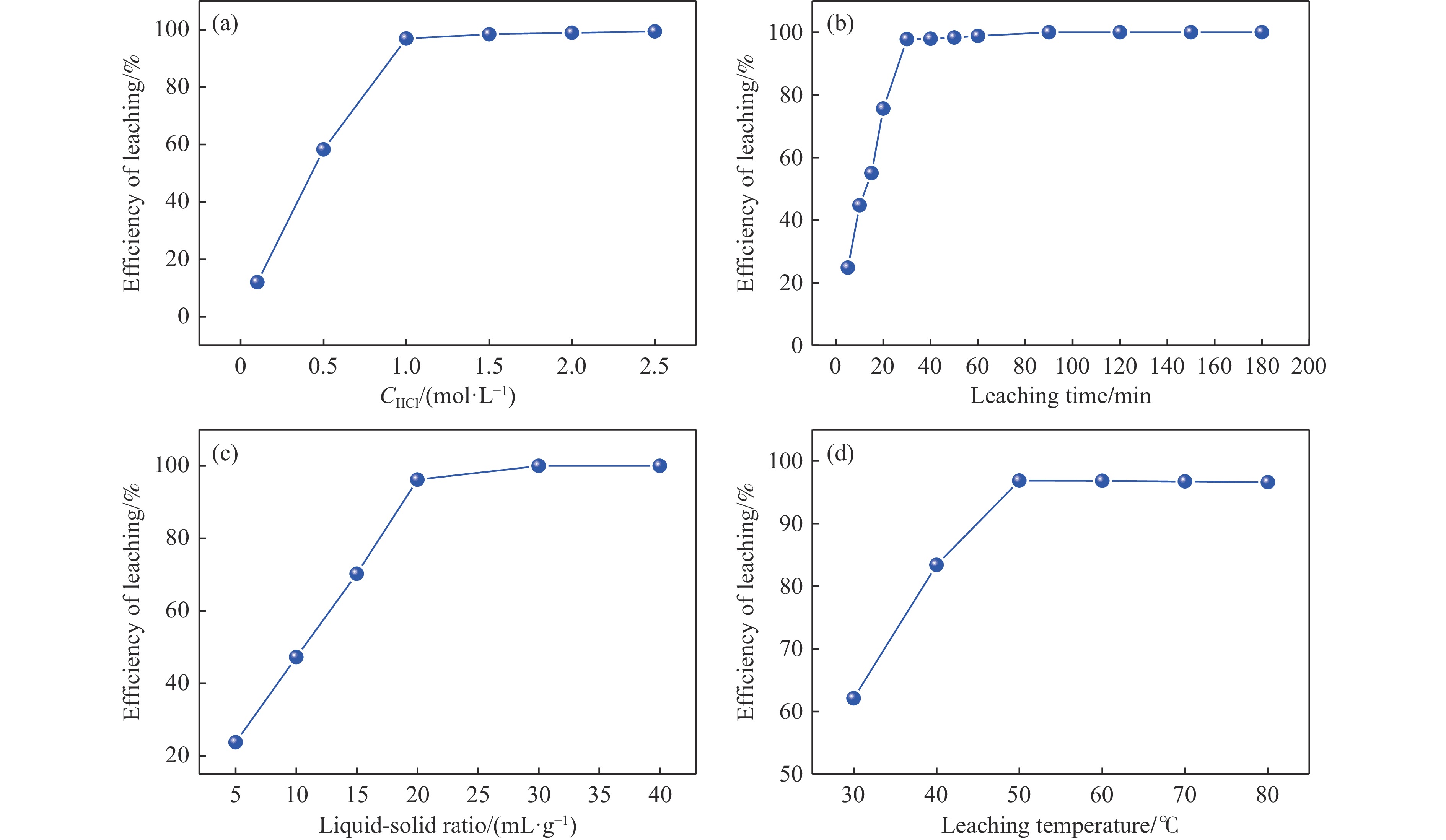
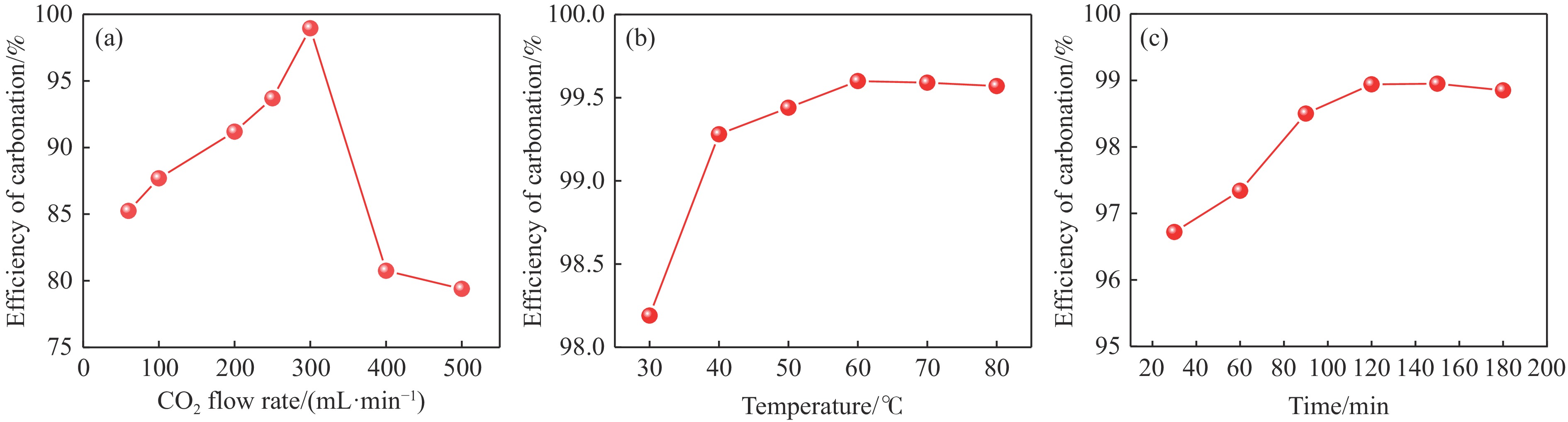
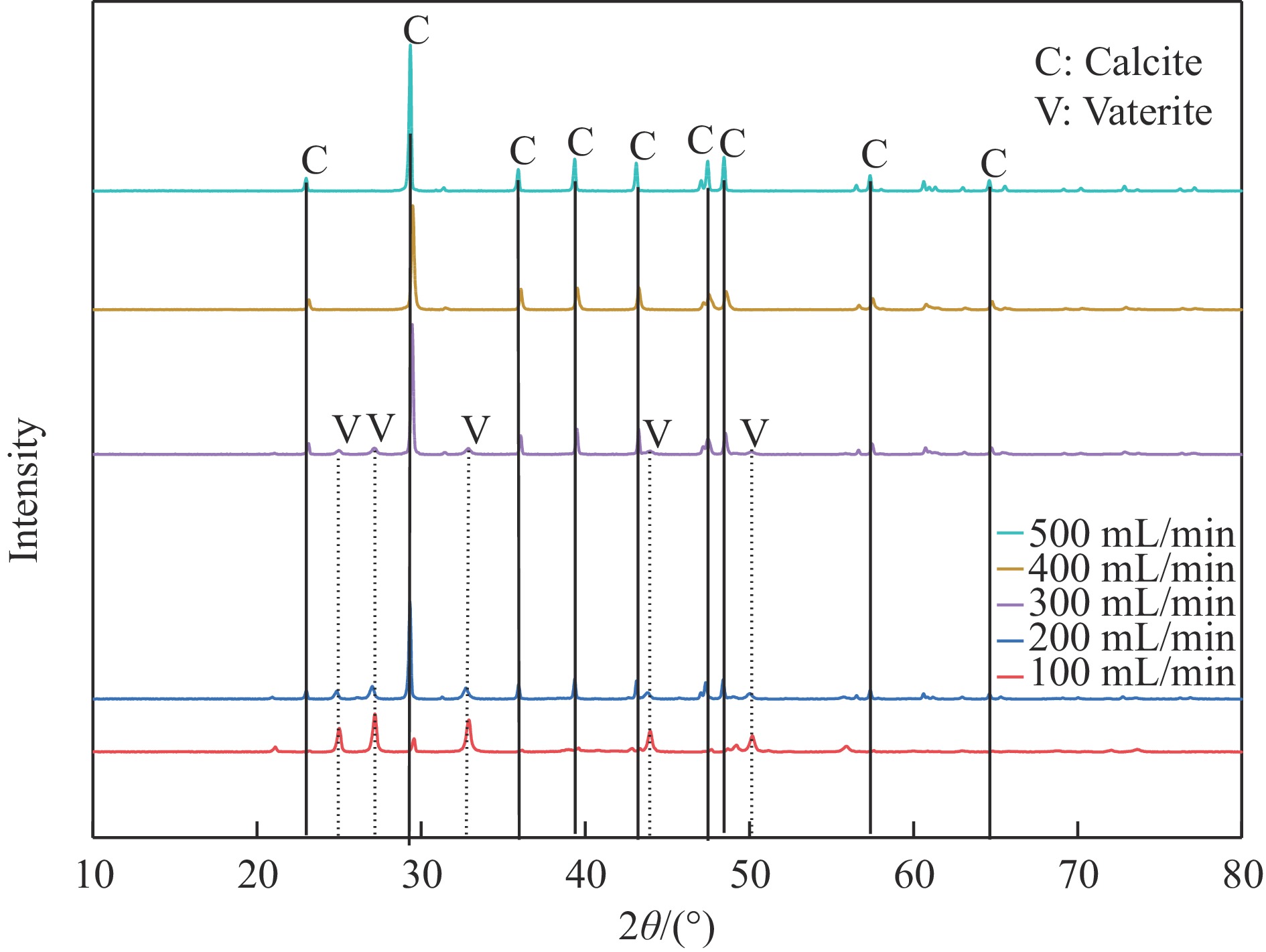
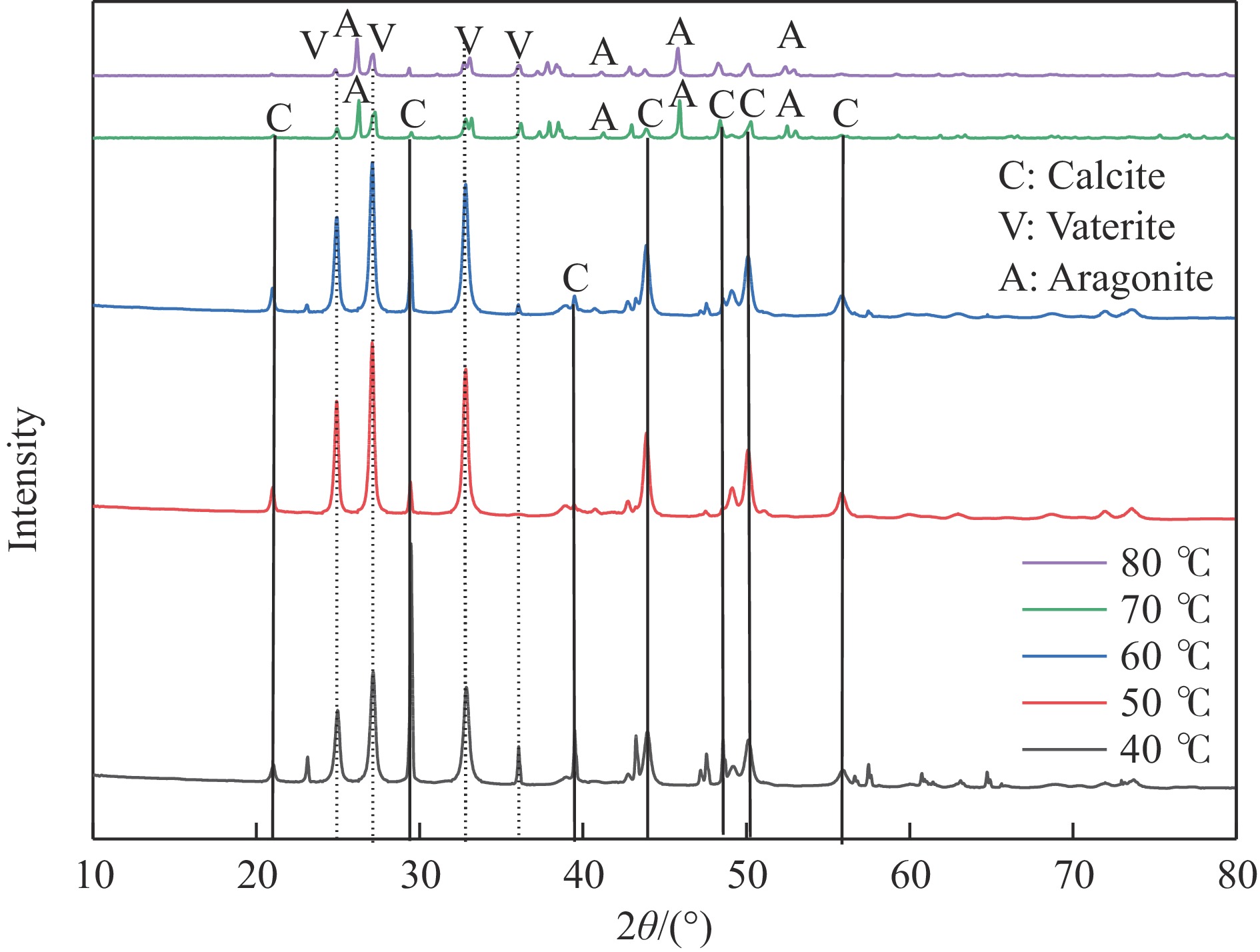
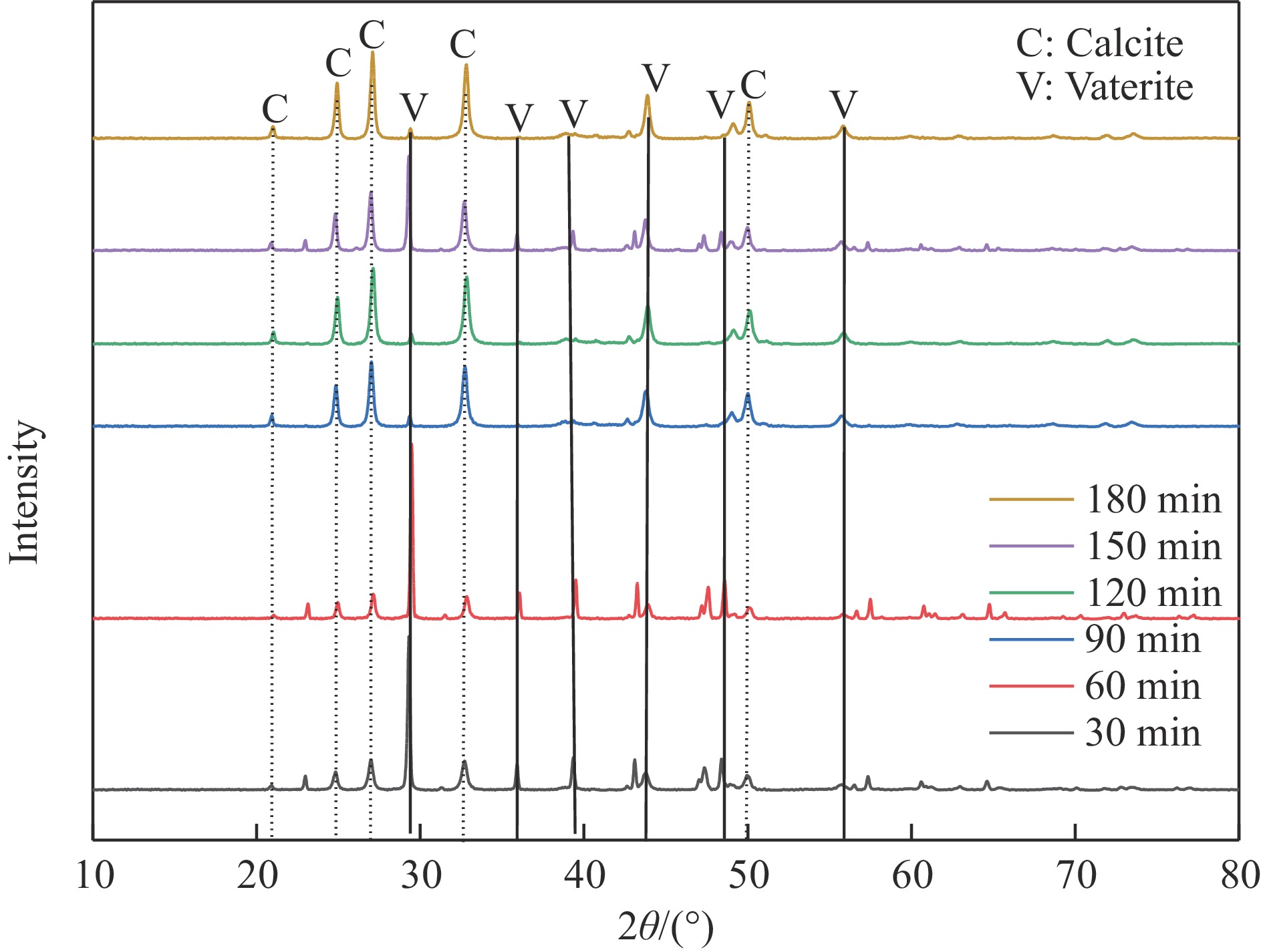
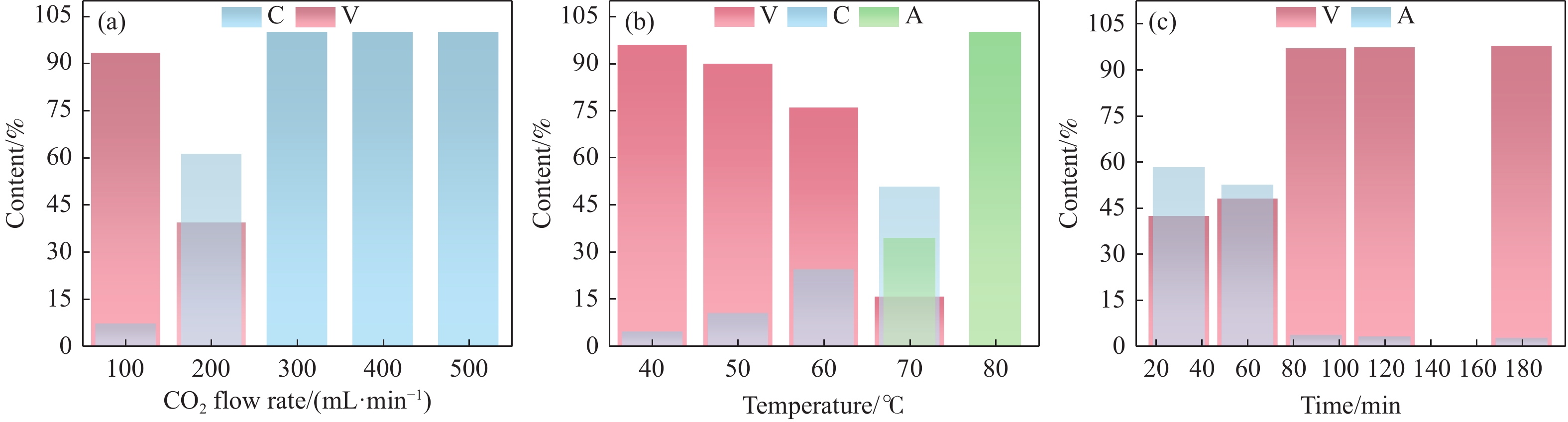
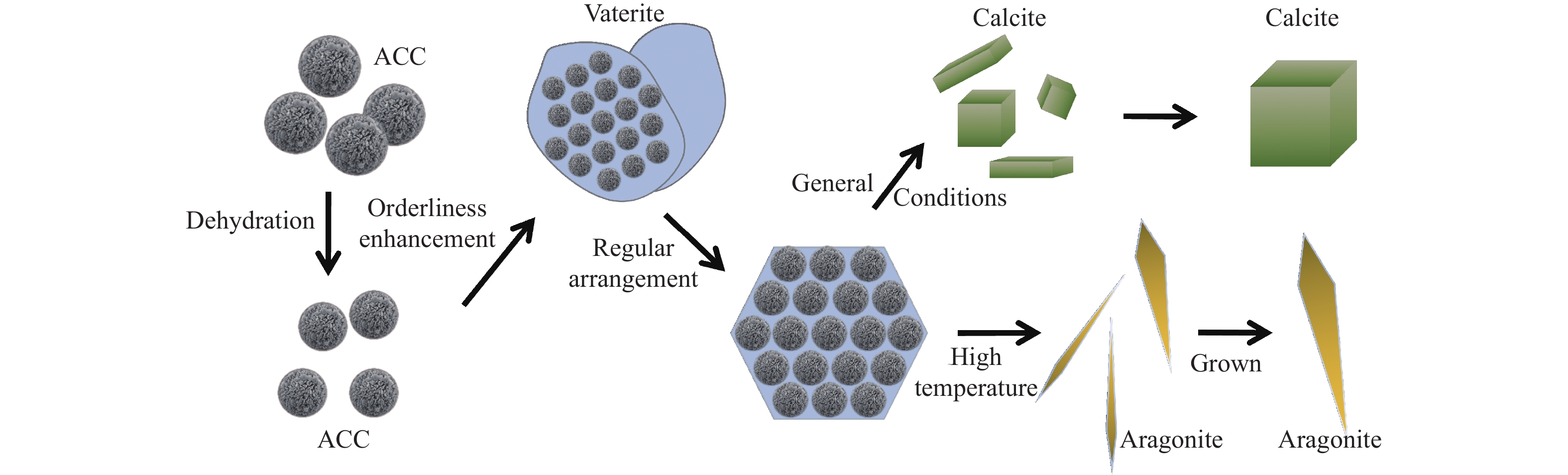
本实验详细研究了浸出剂种类、浓度、反应时间、温度和液固比等对脱碳气化渣中钙浸出率的影响,并讨论了CO2流量、温度、碳酸化时间对碳酸化效率和生成的沉淀碳酸钙(PCC)晶型结构的影响规律。结果表明,在2 mol/L 盐酸、液固比为20 mL/g、反应温度为50 ℃、反应时间为90 min的浸出条件下,钙浸出率最高,为98.79%。在碳酸化阶段,CO2流量主要影响碳酸化效率,通过优化碳酸化反应条件,最高碳酸化效率可达99.59%。而反应温度和时间则会对碳酸钙晶型和形貌产生显著影响,降低反应温度和缩短反应时间更有利于球霰石型碳酸钙的生成。
本实验详细研究了浸出剂种类、浓度、反应时间、温度和液固比等对脱碳气化渣中钙浸出率的影响,并讨论了CO2流量、温度、碳酸化时间对碳酸化效率和生成的沉淀碳酸钙(PCC)晶型结构的影响规律。结果表明,在2 mol/L 盐酸、液固比为20 mL/g、反应温度为50 ℃、反应时间为90 min的浸出条件下,钙浸出率最高,为98.79%。在碳酸化阶段,CO2流量主要影响碳酸化效率,通过优化碳酸化反应条件,最高碳酸化效率可达99.59%。而反应温度和时间则会对碳酸钙晶型和形貌产生显著影响,降低反应温度和缩短反应时间更有利于球霰石型碳酸钙的生成。



当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023078
摘要:
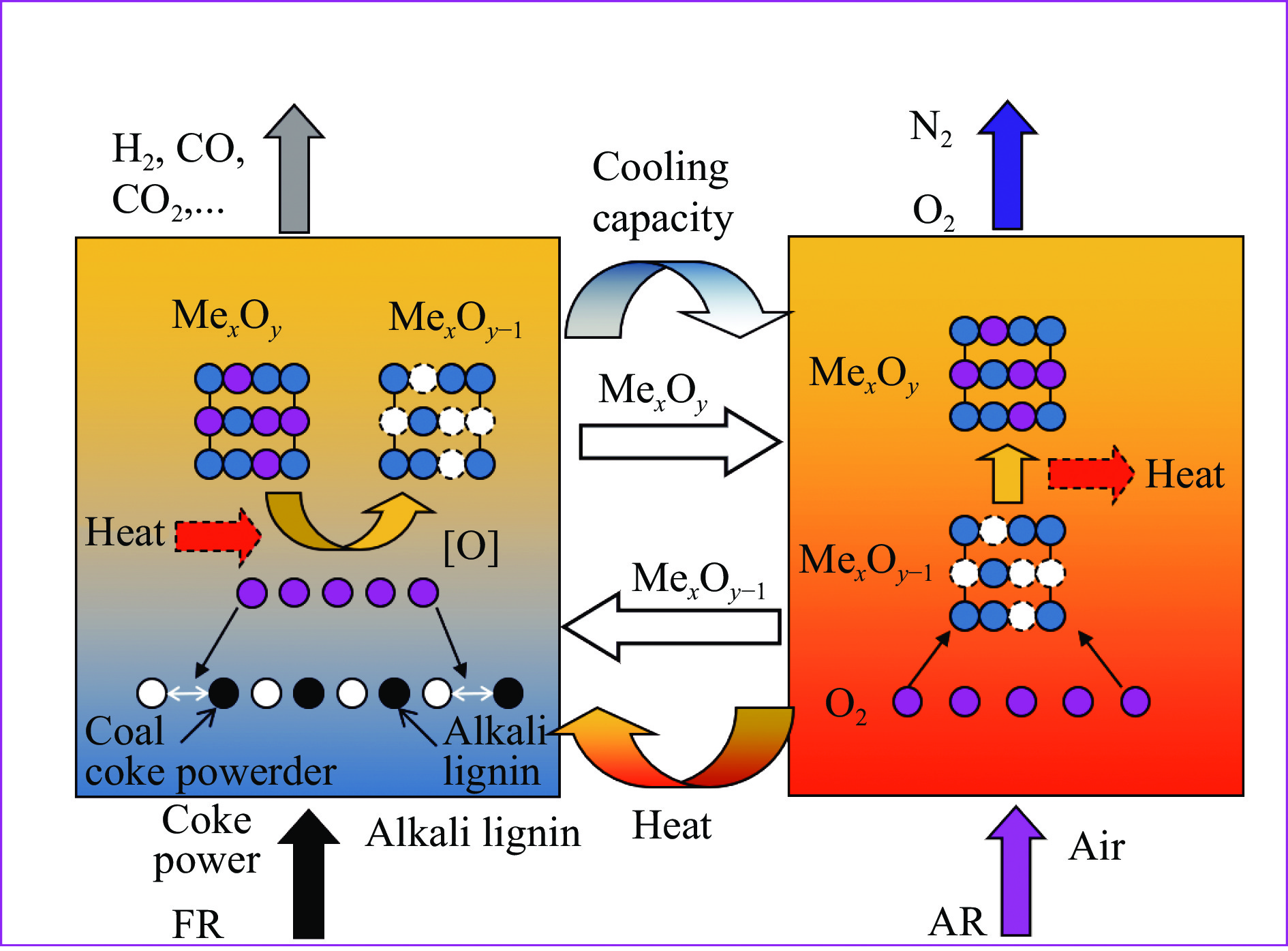
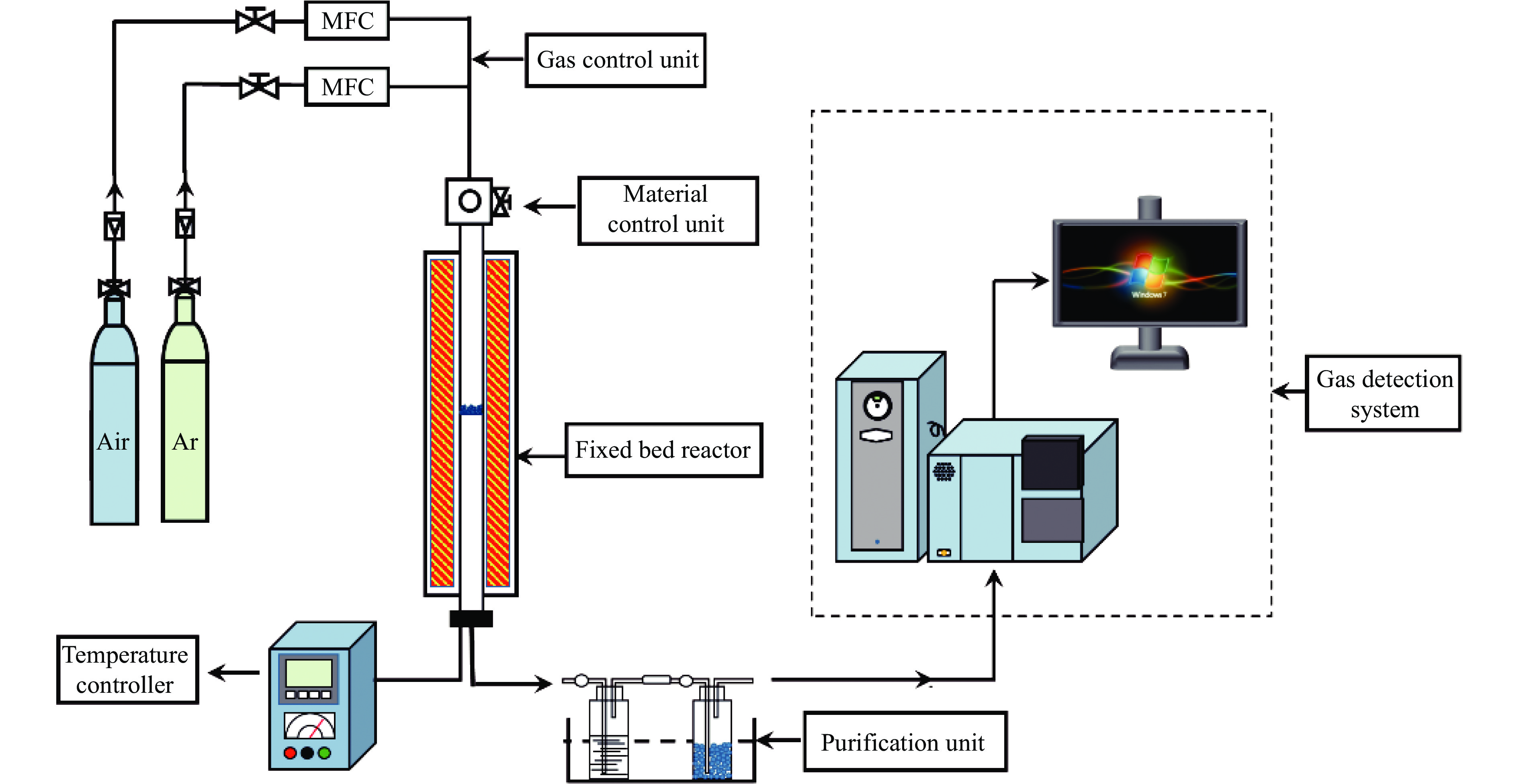
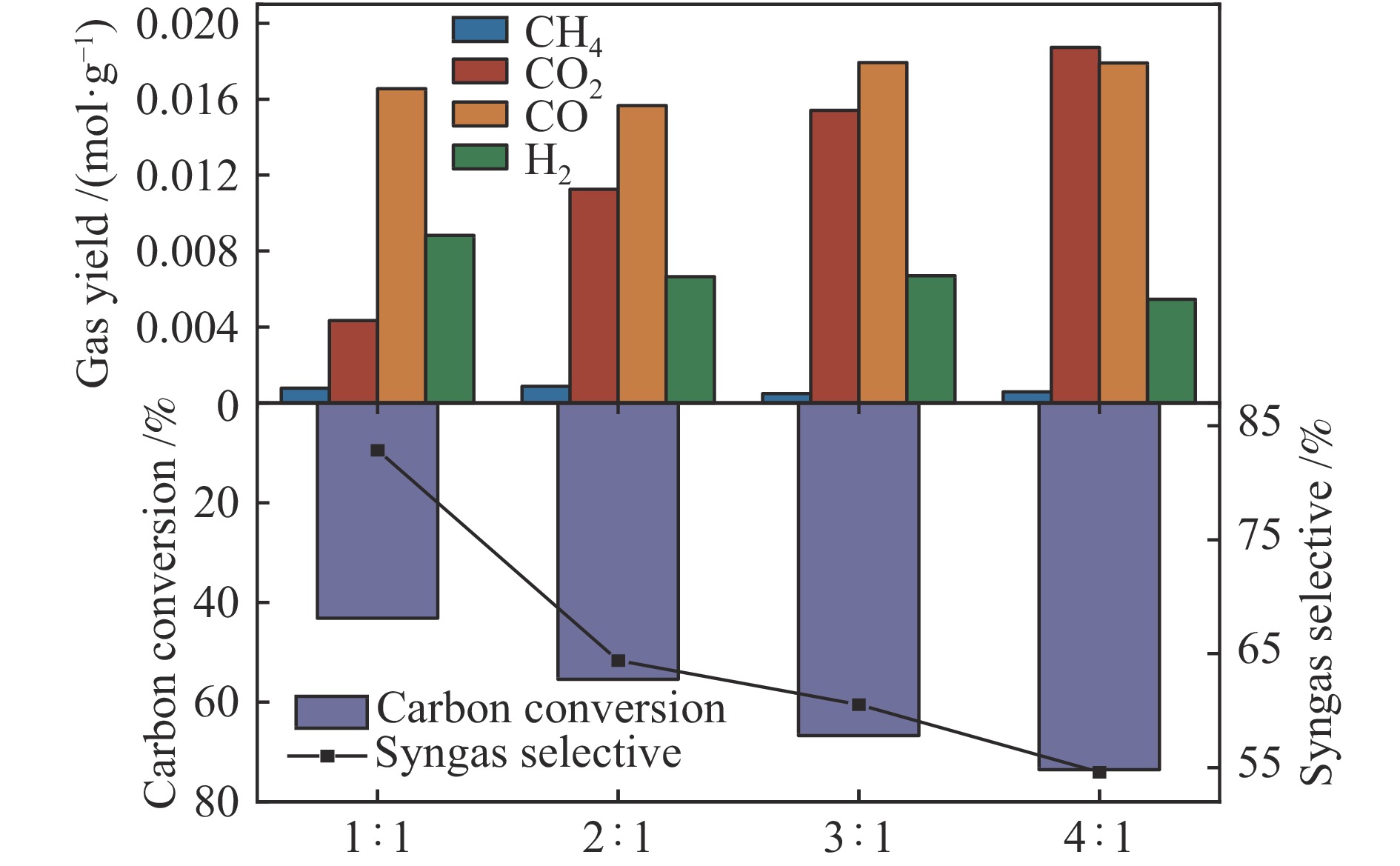
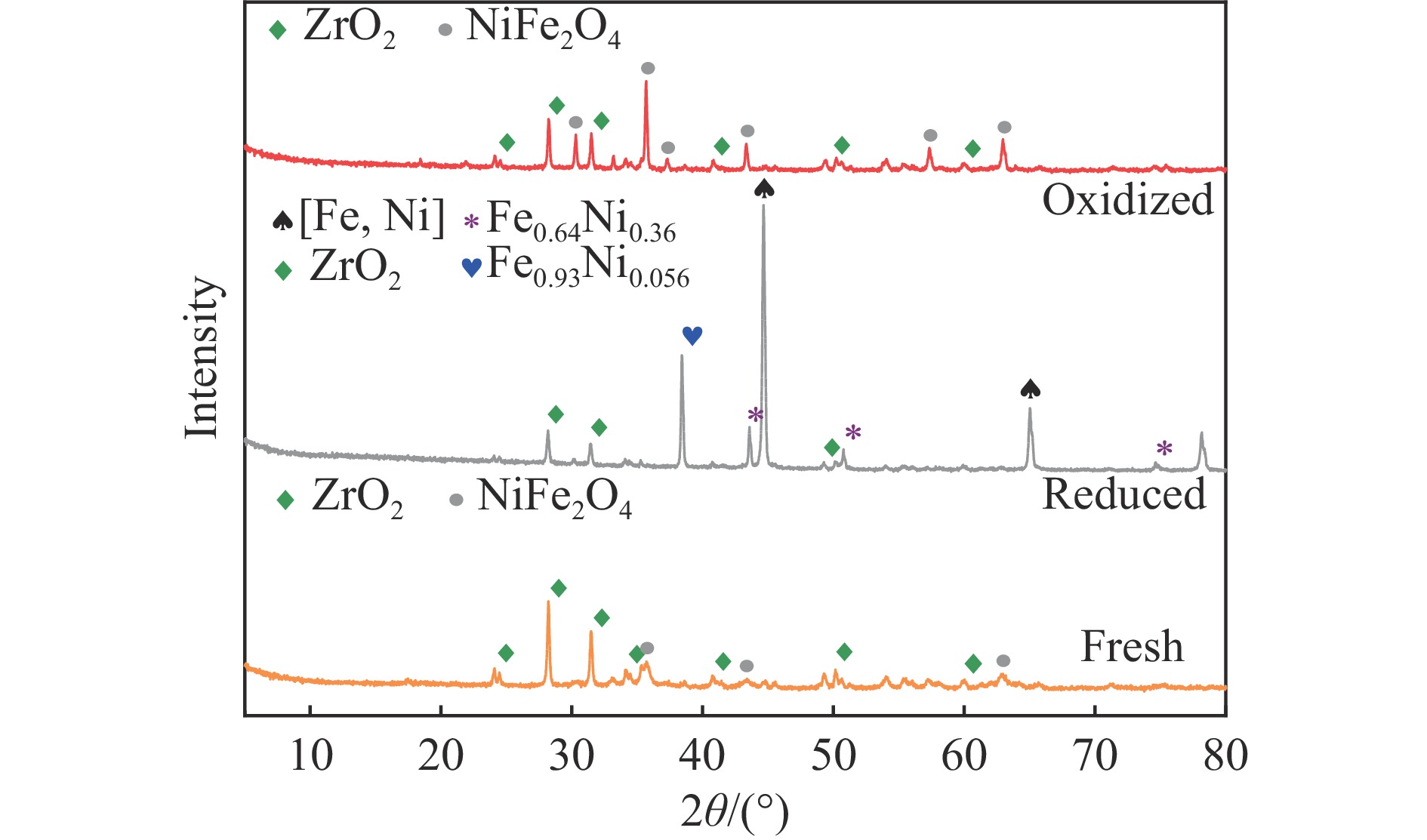
中国煤炼焦工业副产物煤焦粉产量大、活性低,难以被直接回收利用,常规热化学利用方式反应条件苛刻、催化剂易失活且存在动力学限制。本研究通过造纸副产物碱木质素作为可弃型催化剂,构建碱木质素强化化学链气化的方式来处理煤焦粉,实现工业副产物协同资源化利用。热转化实验和动力学分析研究表明,碱木质素可强化煤焦粉化学链气化过程,促进煤焦粉热解峰向低温方向移动。当煤焦粉与碱木质素质量比为1∶3时,反应活化能比单独煤焦粉反应降低87.56%。固定床实验证实气化温度提高、碱木质素以及载氧体赋存量增加,可以有效提高燃料碳转化率及合成气产物择性,促进气化反应进行,但氧载体过量会导致合成气转化为终端产物,降低合成气选择性。在气化温度为950 ℃,煤焦粉与碱木质素质量比为1∶2, 氧载体与煤焦粉/碱木质素混合体系质量比为1∶1的最佳反应条件下,基于NiFe2O3的碱木质素/煤焦粉化学链气化合成气选择性高达82.85%。该研究为碱木质素与煤焦粉的资源化利用提供科学依据。
中国煤炼焦工业副产物煤焦粉产量大、活性低,难以被直接回收利用,常规热化学利用方式反应条件苛刻、催化剂易失活且存在动力学限制。本研究通过造纸副产物碱木质素作为可弃型催化剂,构建碱木质素强化化学链气化的方式来处理煤焦粉,实现工业副产物协同资源化利用。热转化实验和动力学分析研究表明,碱木质素可强化煤焦粉化学链气化过程,促进煤焦粉热解峰向低温方向移动。当煤焦粉与碱木质素质量比为1∶3时,反应活化能比单独煤焦粉反应降低87.56%。固定床实验证实气化温度提高、碱木质素以及载氧体赋存量增加,可以有效提高燃料碳转化率及合成气产物择性,促进气化反应进行,但氧载体过量会导致合成气转化为终端产物,降低合成气选择性。在气化温度为950 ℃,煤焦粉与碱木质素质量比为1∶2, 氧载体与煤焦粉/碱木质素混合体系质量比为1∶1的最佳反应条件下,基于NiFe2O3的碱木质素/煤焦粉化学链气化合成气选择性高达82.85%。该研究为碱木质素与煤焦粉的资源化利用提供科学依据。
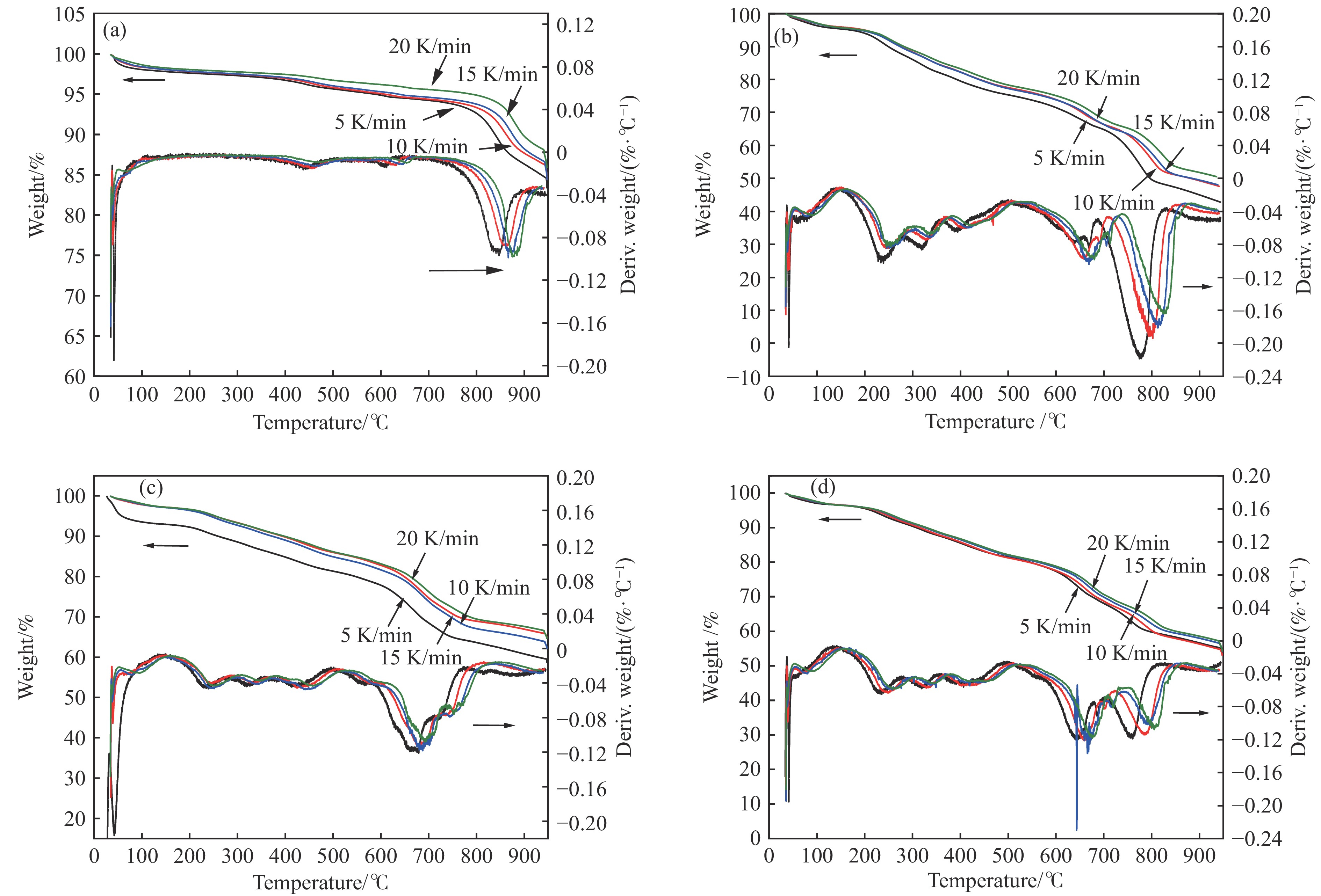
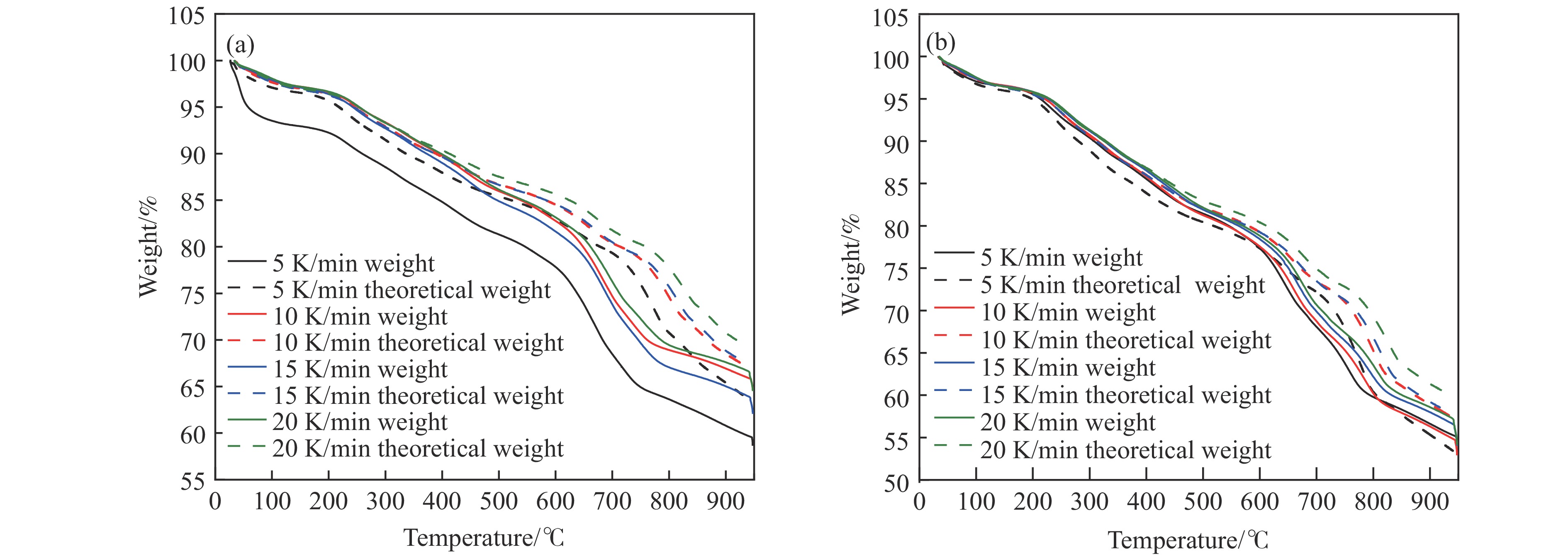
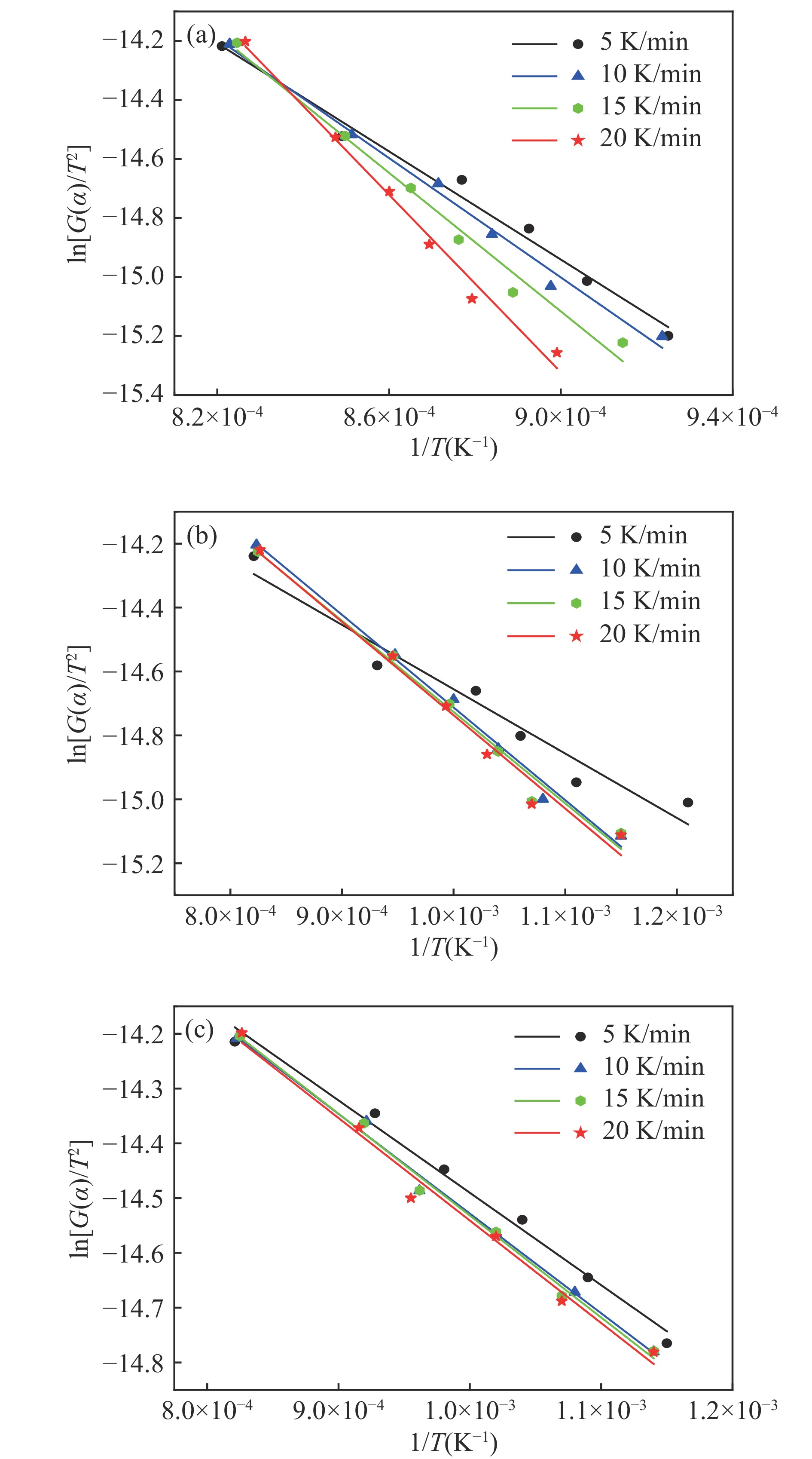

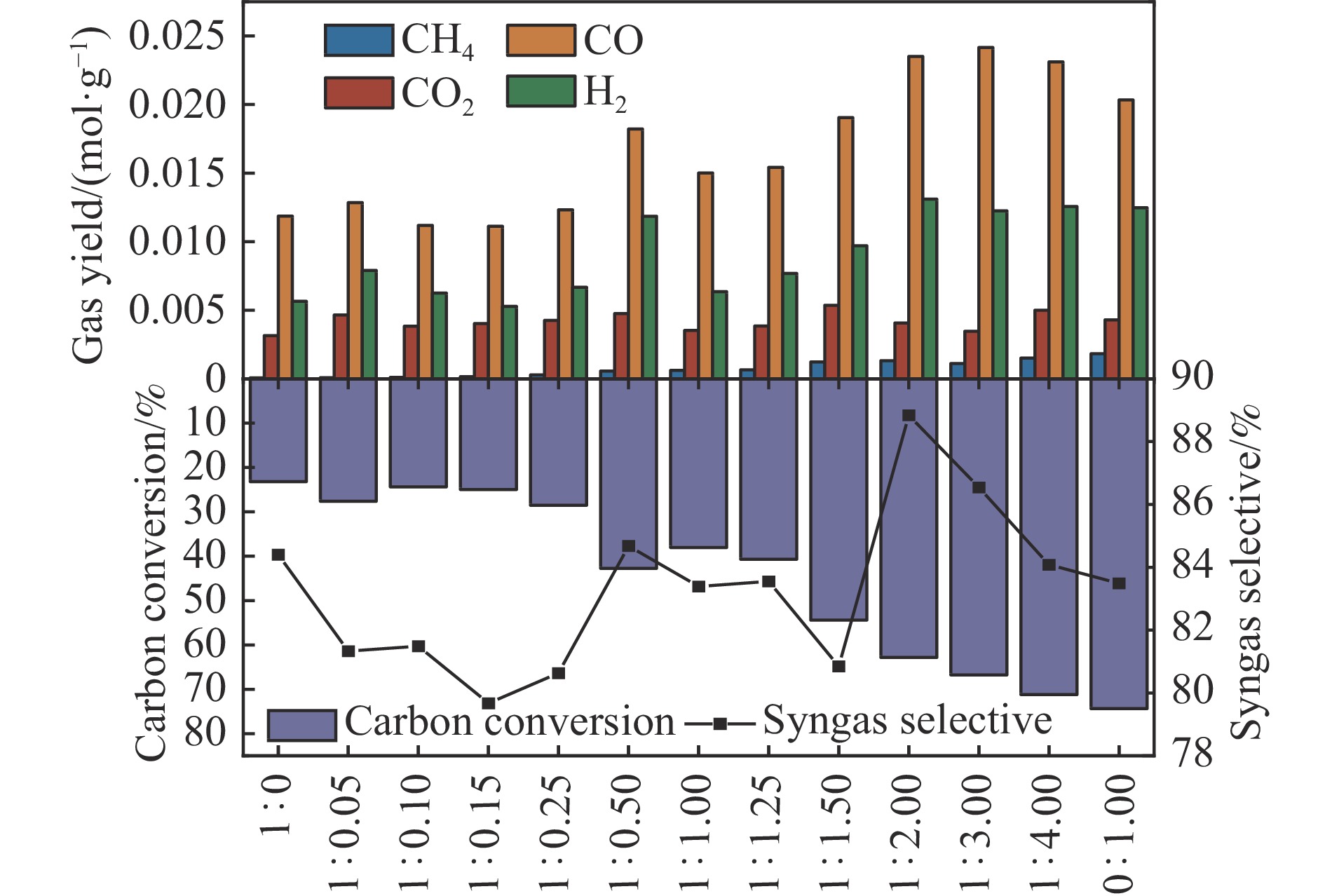

当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60434-2
摘要:
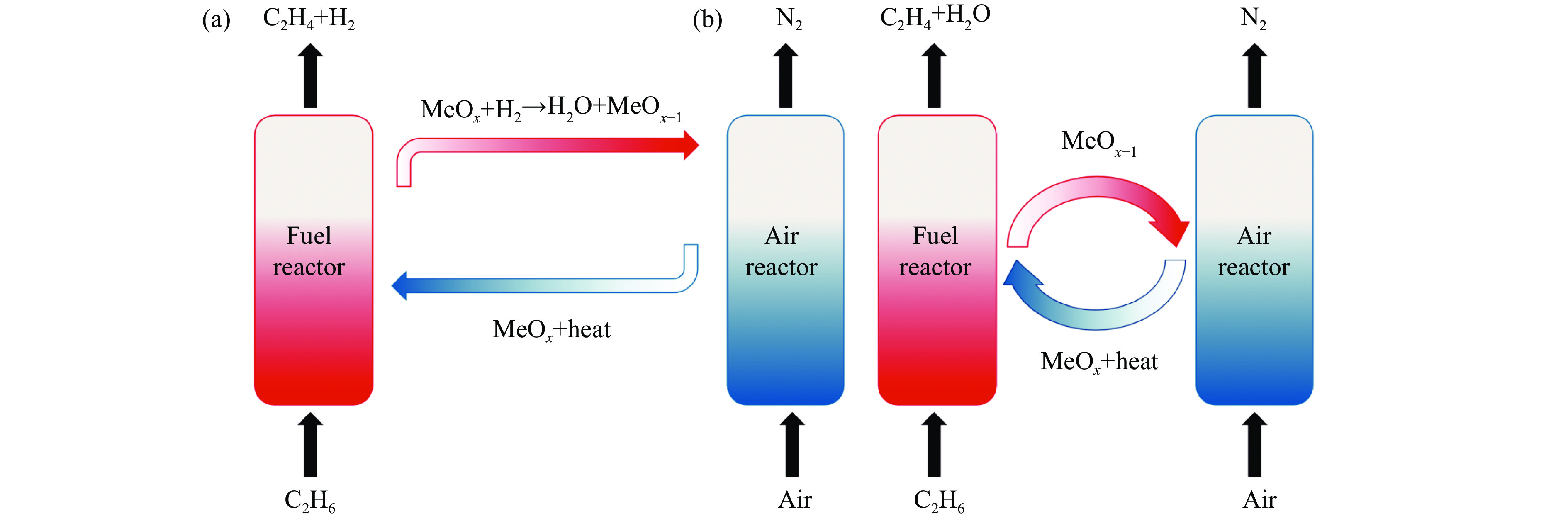
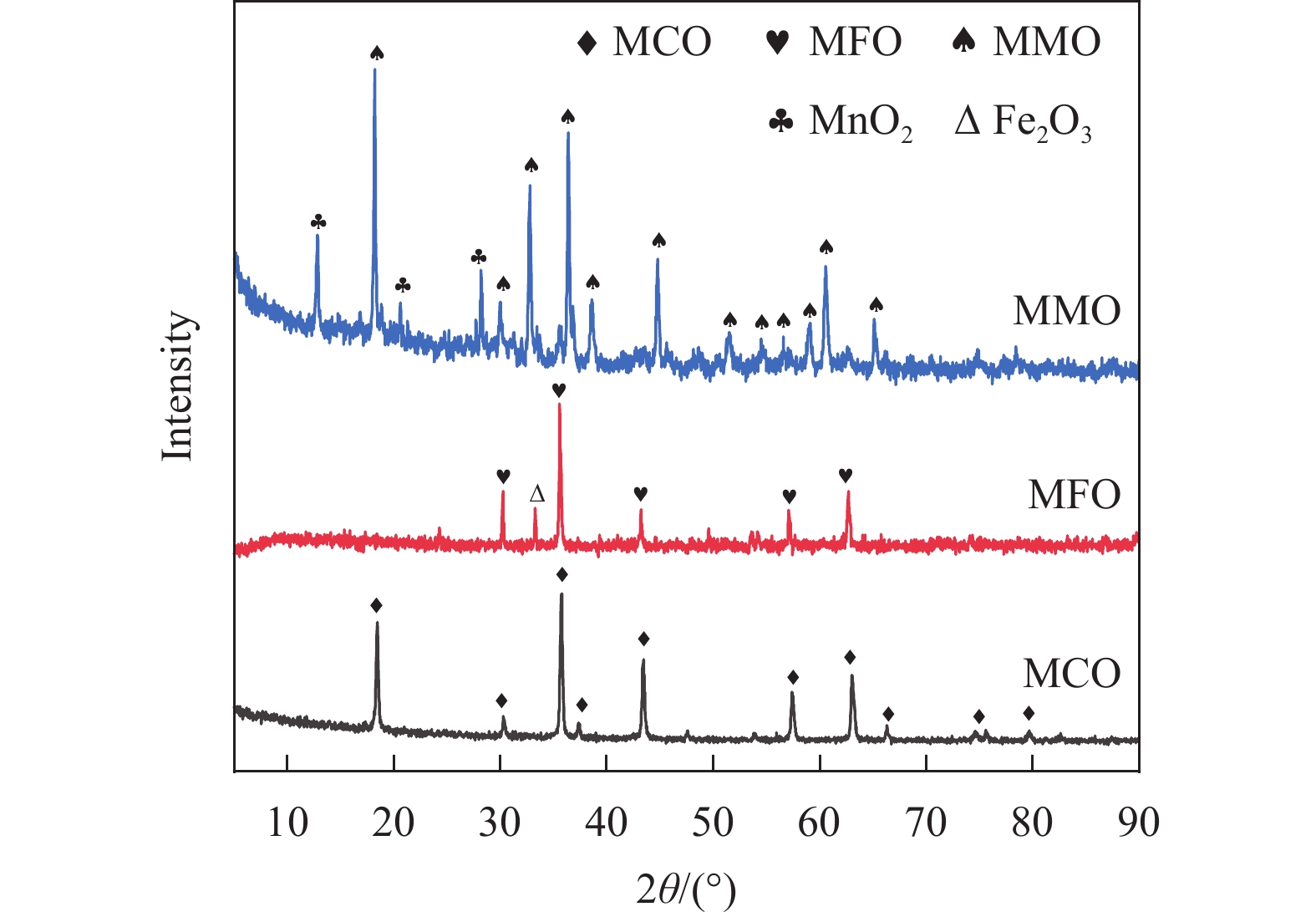
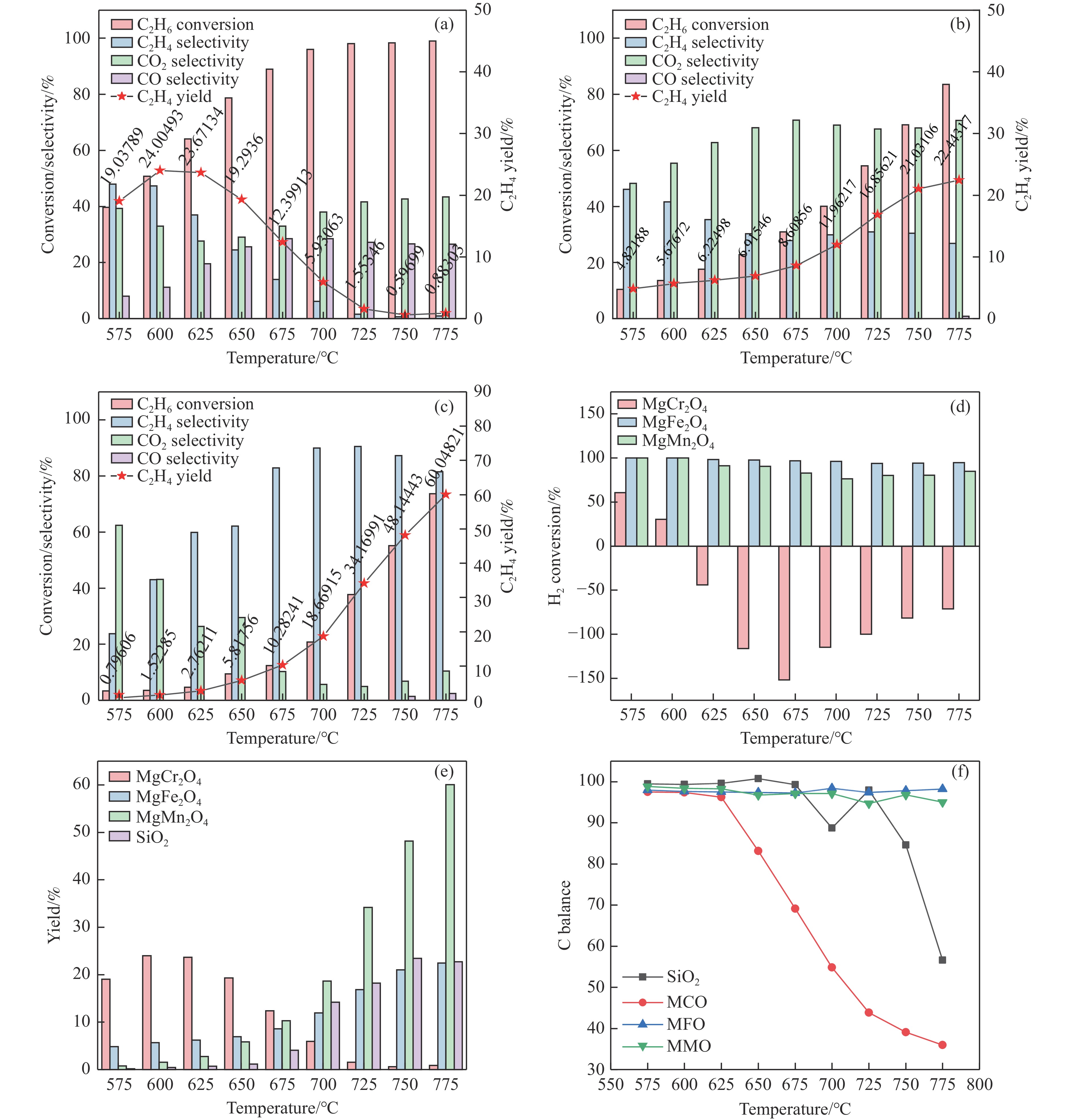
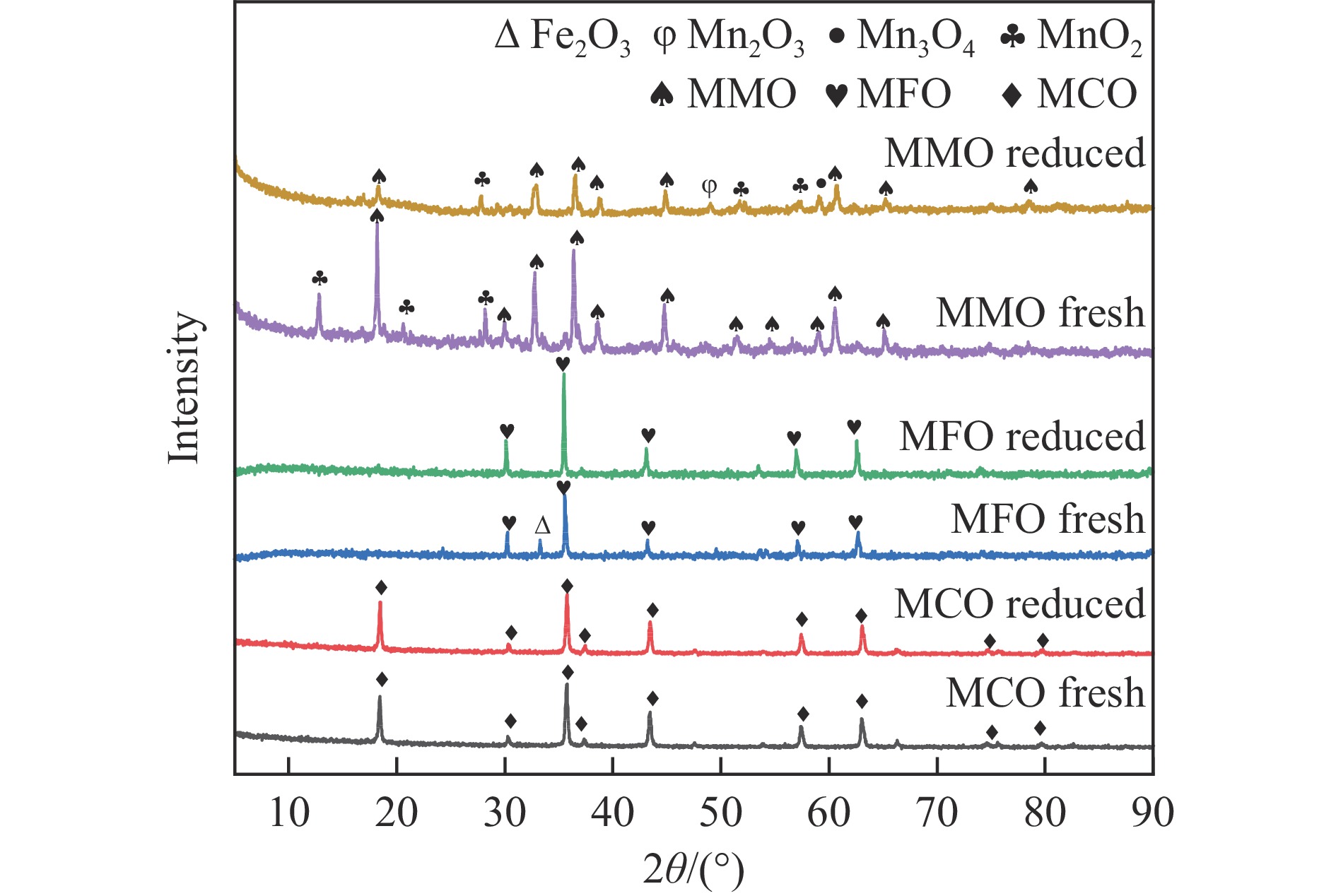
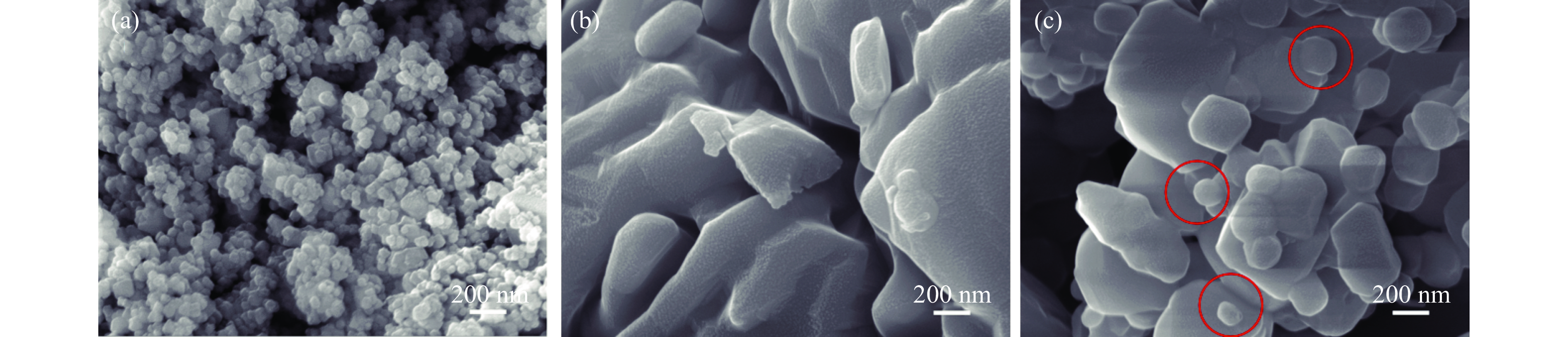
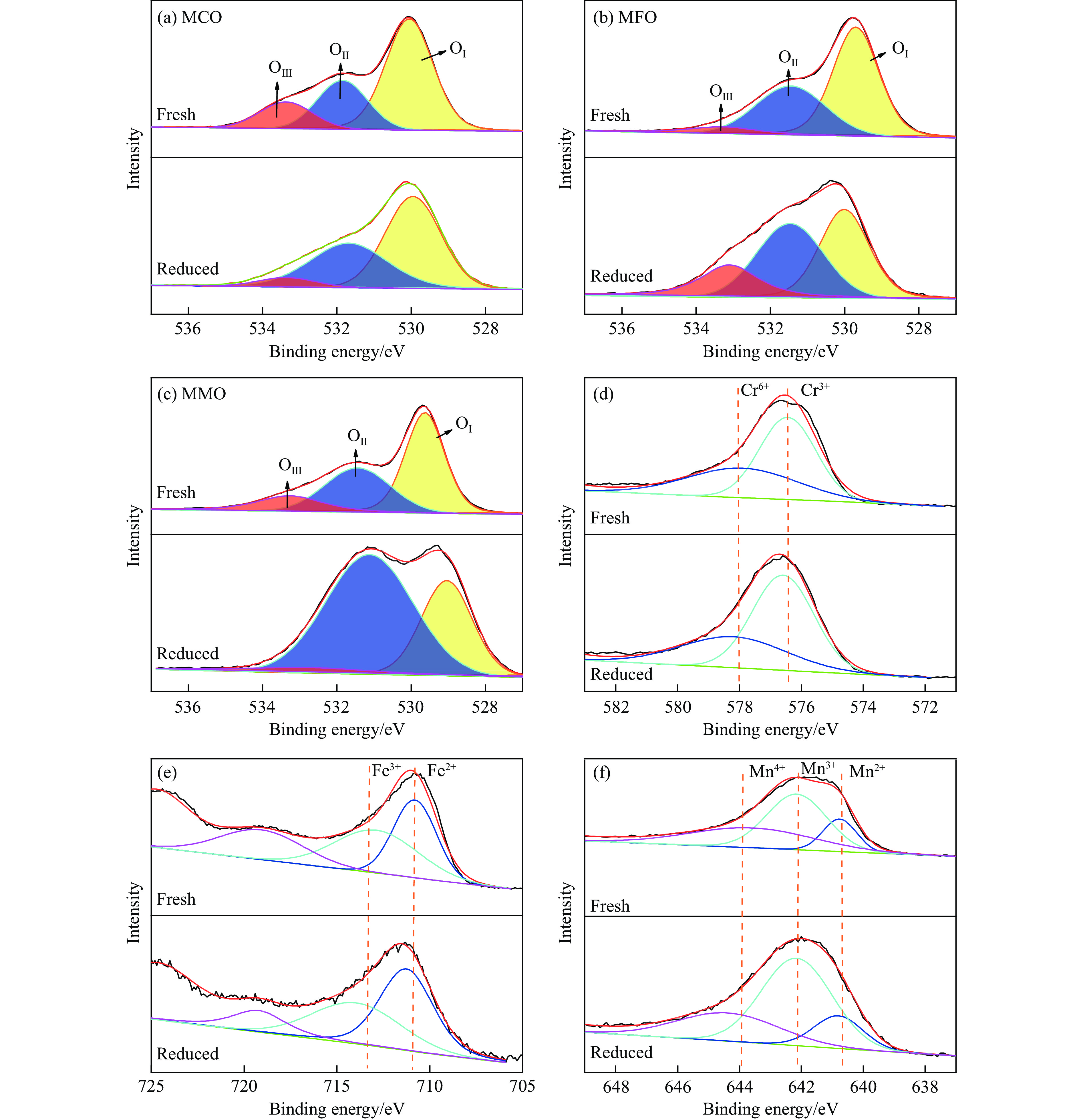
Chemical looping oxidative dehydrogenation (CL-ODH) provides a multifunctional conversion platform that can take advantage of the selective oxidation of lattice oxygen in oxygen carrier to achieve high-valued ethane to ethylene conversion. In this study, we explored the effect of B-site element in MgX2O4 (X=Cr, Fe, or Mn) spinel-type oxygen carriers on the performance of ethane CL-ODH. The properties test and characterization of MgX2O4 spinel were tested by fixed bed and H2-TPR, O2-TPD, TG, in situ Raman, SEM, and TEM. The results showed that because MgCr2O4 only released a small amount of adsorbed surface oxygen, it tended to catalyze the conversion of ethane to coke and hydrogen. MgFe2O4 facilitated the deep oxidation of ethane into CO2 by providing more surface lattice oxygen. Meanwhile, since a significant amount of bulk lattice oxygen was released by the MgMn2O4 oxygen carrier, it could burn hydrogen in a targeted manner to advance the reaction and increased ethylene's selectivity. Thereby, MgMn2O4 achieved an ethane conversion of 73.7% with an ethylene selectivity of 81.46%. Furthermore, the MgMn2O4 catalyst demonstrated stable reactivity and an ethylene yield of about 62% in ethane CL-ODH over the 30 redox cycles. The screening tests indicated that the B-site elements in MgX2O4 spinel oxides could significantly influence their ability to supply lattice oxygen, thereby affecting their performance in ethane CL-ODH reaction.
Chemical looping oxidative dehydrogenation (CL-ODH) provides a multifunctional conversion platform that can take advantage of the selective oxidation of lattice oxygen in oxygen carrier to achieve high-valued ethane to ethylene conversion. In this study, we explored the effect of B-site element in MgX2O4 (X=Cr, Fe, or Mn) spinel-type oxygen carriers on the performance of ethane CL-ODH. The properties test and characterization of MgX2O4 spinel were tested by fixed bed and H2-TPR, O2-TPD, TG, in situ Raman, SEM, and TEM. The results showed that because MgCr2O4 only released a small amount of adsorbed surface oxygen, it tended to catalyze the conversion of ethane to coke and hydrogen. MgFe2O4 facilitated the deep oxidation of ethane into CO2 by providing more surface lattice oxygen. Meanwhile, since a significant amount of bulk lattice oxygen was released by the MgMn2O4 oxygen carrier, it could burn hydrogen in a targeted manner to advance the reaction and increased ethylene's selectivity. Thereby, MgMn2O4 achieved an ethane conversion of 73.7% with an ethylene selectivity of 81.46%. Furthermore, the MgMn2O4 catalyst demonstrated stable reactivity and an ethylene yield of about 62% in ethane CL-ODH over the 30 redox cycles. The screening tests indicated that the B-site elements in MgX2O4 spinel oxides could significantly influence their ability to supply lattice oxygen, thereby affecting their performance in ethane CL-ODH reaction.


当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023081
摘要:
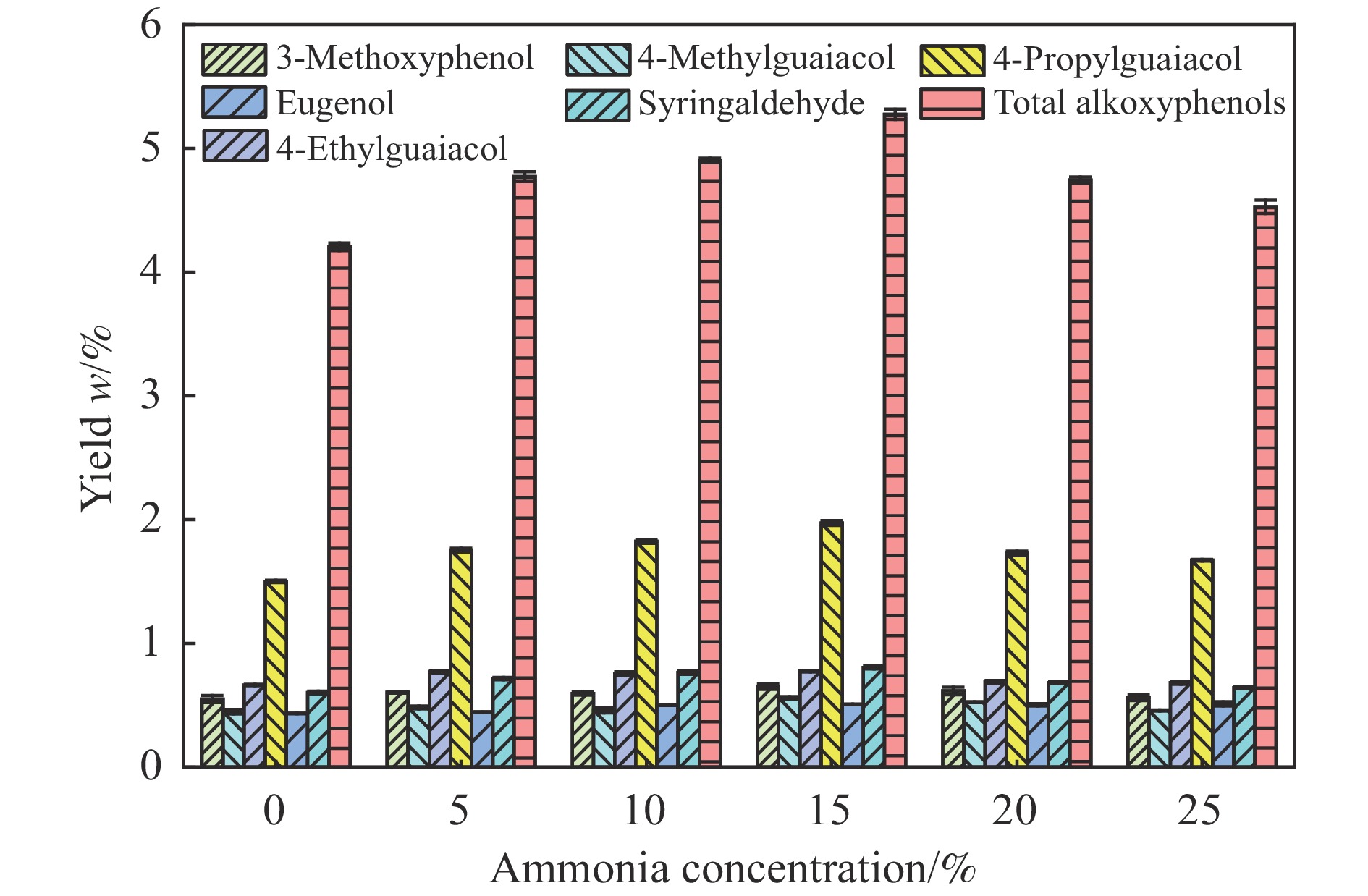
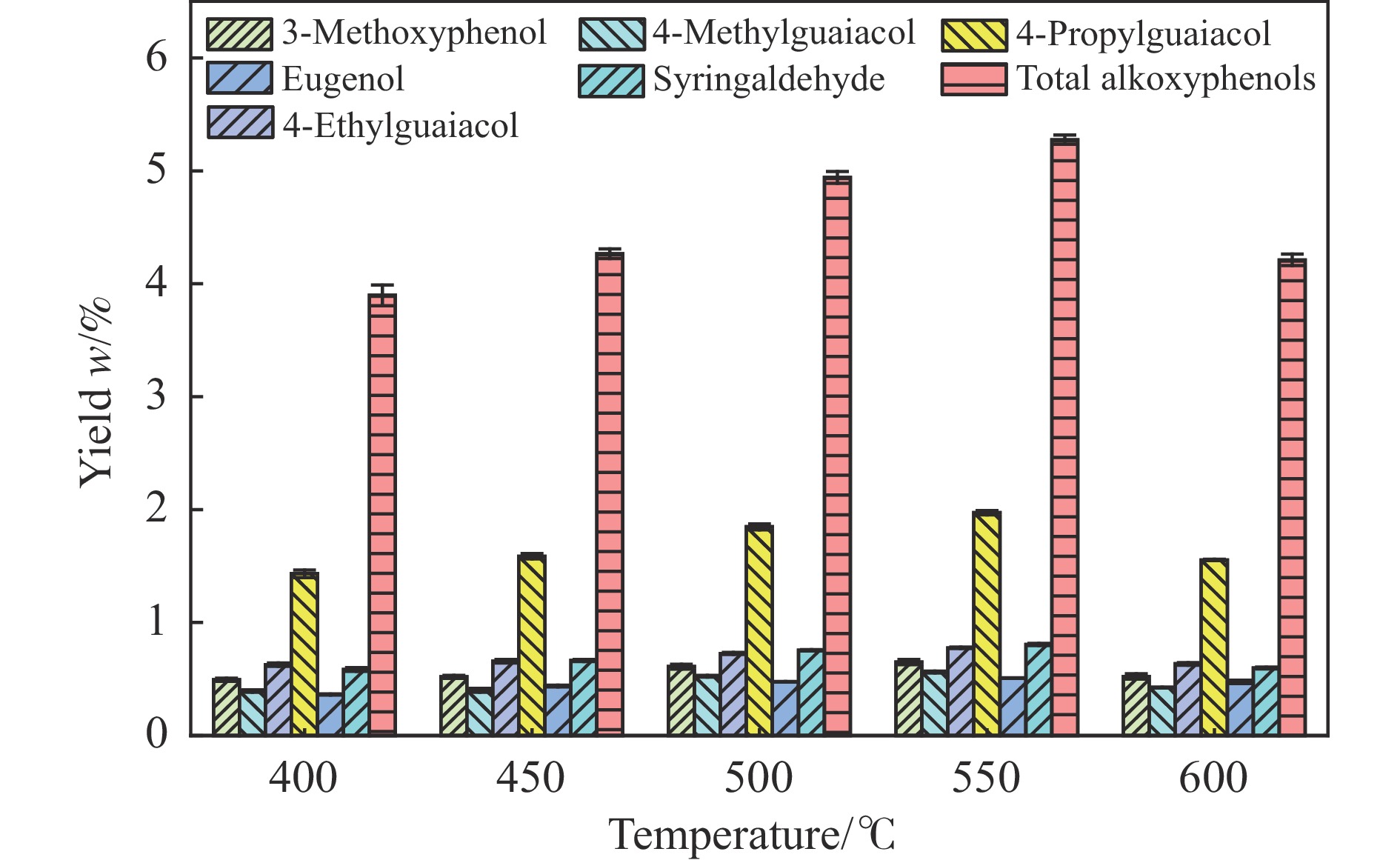
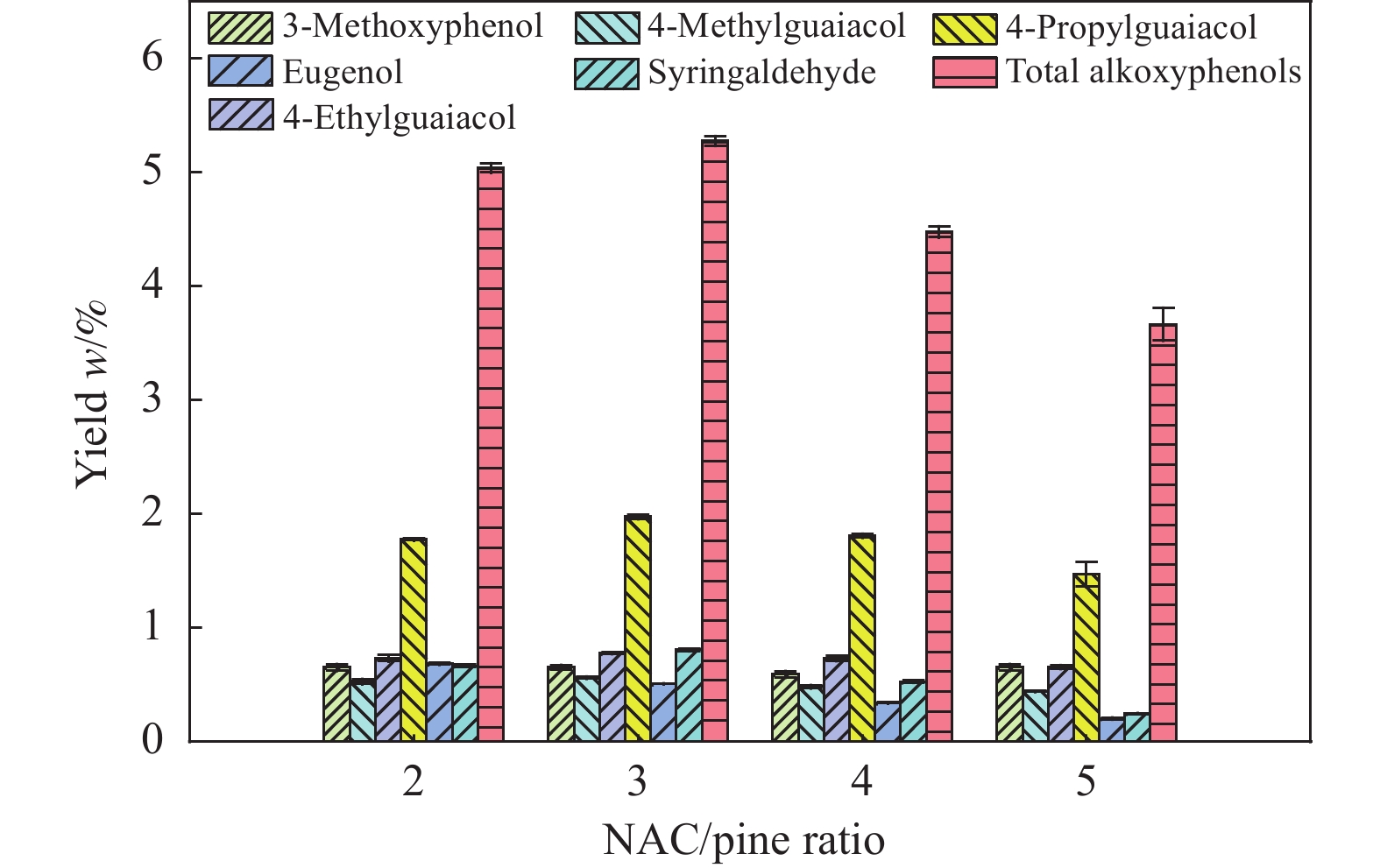
本研究以核桃壳基掺氮活性炭(NAC)为催化剂、以9,10-二氢蒽(DHA)为供氢剂,开展了松木选择性热解制备烷氧基酚研究,探究了氨水浓度对NAC理化性能的影响,揭示了DHA/松木比、热解温度、NAC/松木比对烷氧基酚生成的调控机制。结果表明,合适的氨水浓度能够改善NAC孔隙结构及活性位点分布,当氨水浓度为15%时,所制备NAC对烷氧基酚的生成促进效果最佳;当DHA/松木比为3、热解温度为550 ℃、NAC/松木比为3时,烷氧基酚产率最大,为5.27%,明显高于纯松木直接催化热解时烷氧基酚产率(1.74%)。
本研究以核桃壳基掺氮活性炭(NAC)为催化剂、以9,10-二氢蒽(DHA)为供氢剂,开展了松木选择性热解制备烷氧基酚研究,探究了氨水浓度对NAC理化性能的影响,揭示了DHA/松木比、热解温度、NAC/松木比对烷氧基酚生成的调控机制。结果表明,合适的氨水浓度能够改善NAC孔隙结构及活性位点分布,当氨水浓度为15%时,所制备NAC对烷氧基酚的生成促进效果最佳;当DHA/松木比为3、热解温度为550 ℃、NAC/松木比为3时,烷氧基酚产率最大,为5.27%,明显高于纯松木直接催化热解时烷氧基酚产率(1.74%)。



当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023076
摘要:
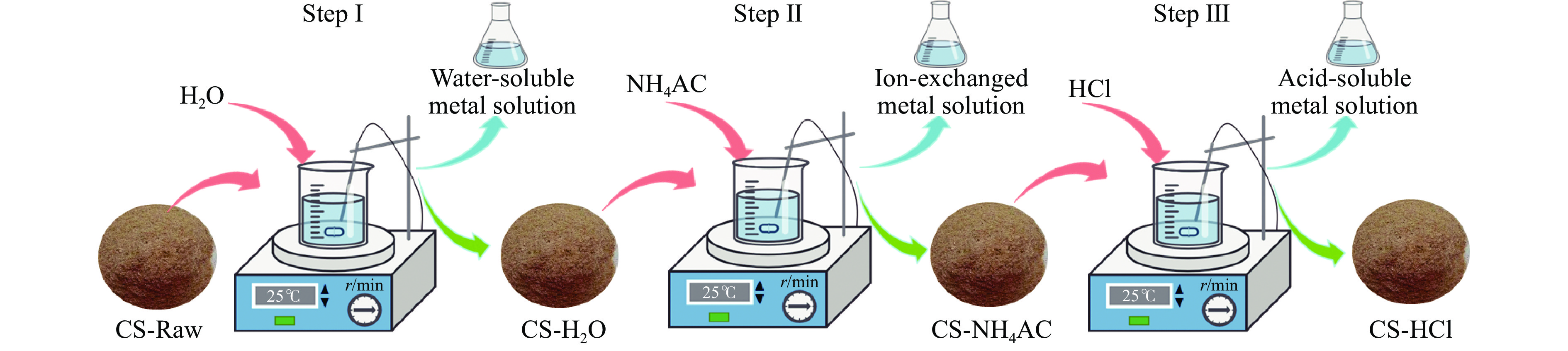
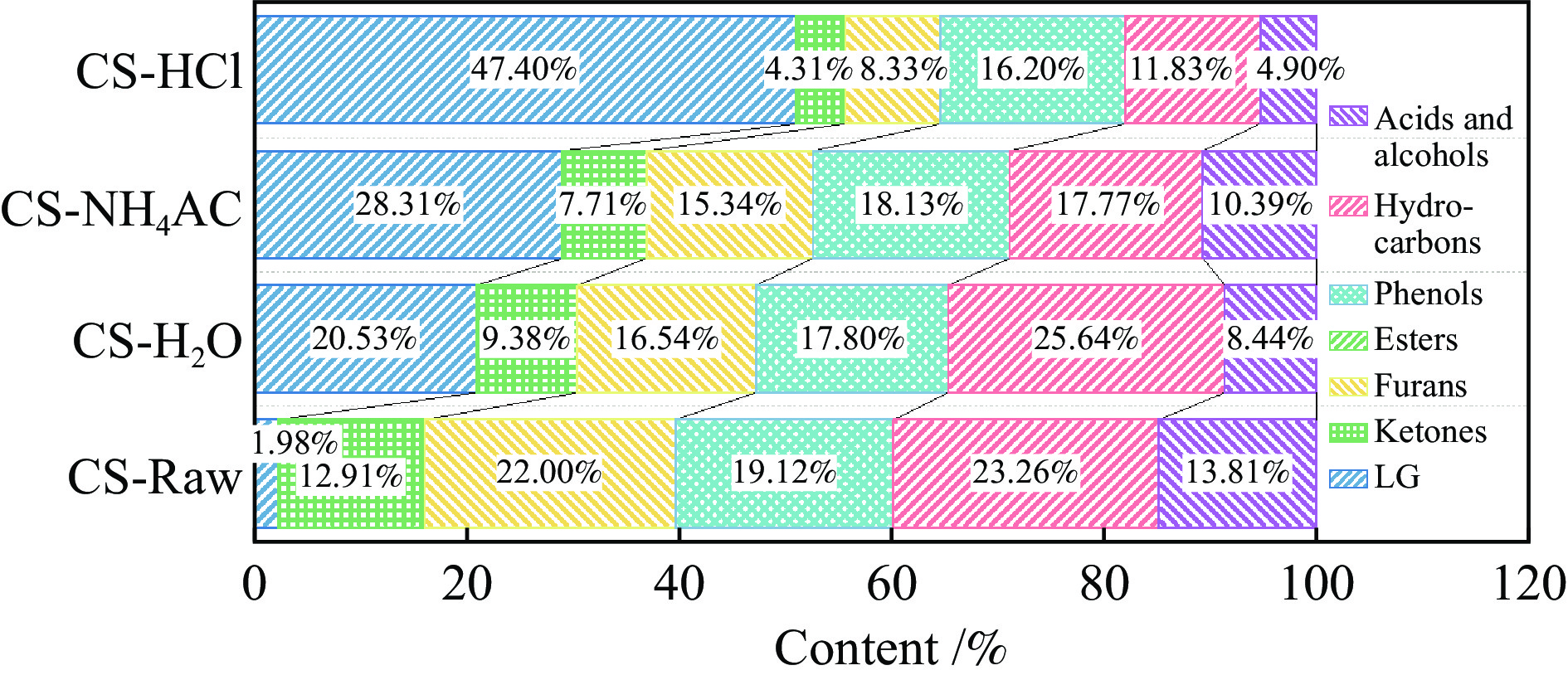
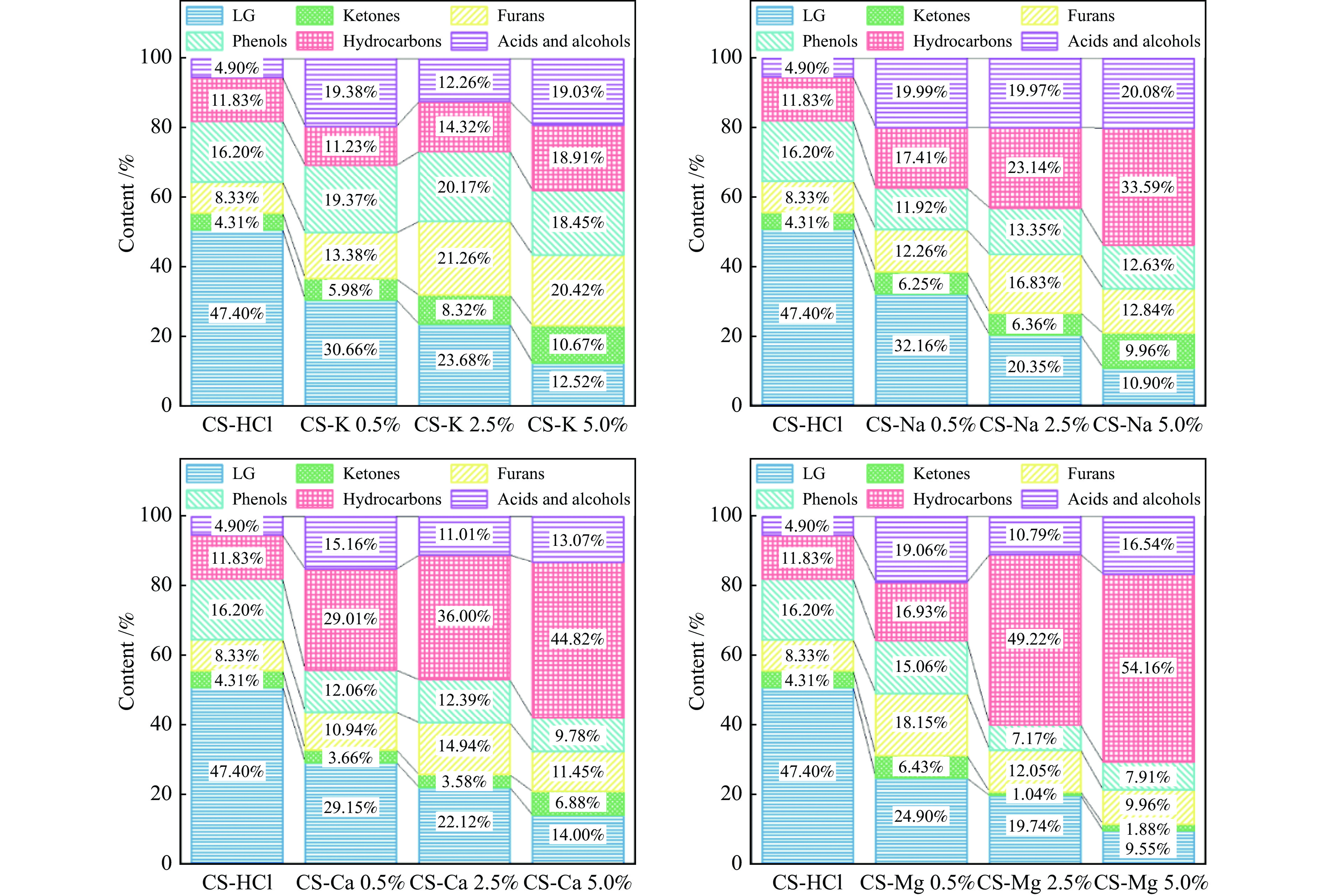
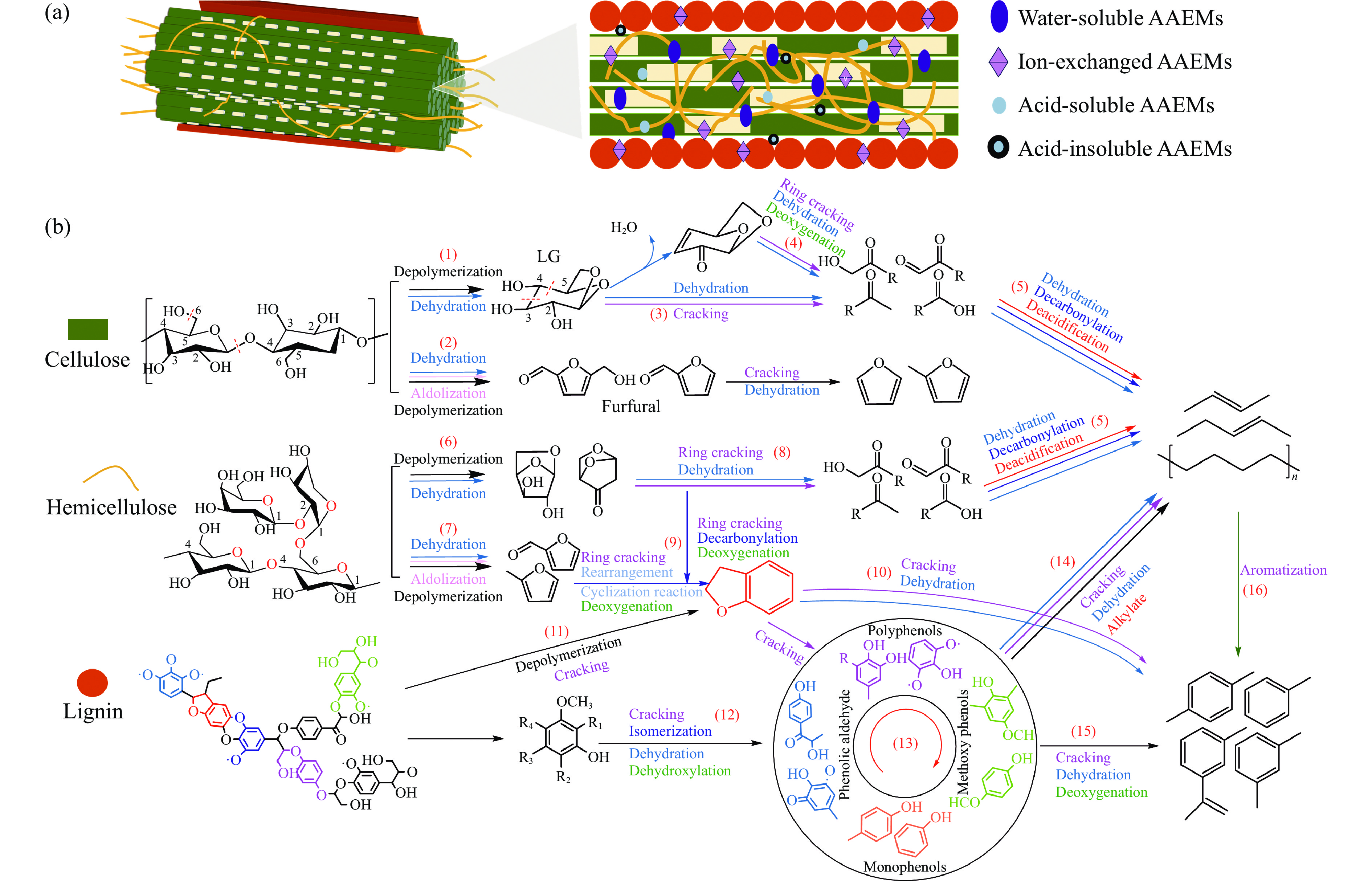
生物质灰分中的碱和碱土金属(AAEMs)对快速热解生物油的产率和组分分布具有显著影响。本研究选取玉米秸秆为原料,研究梯级脱灰预处理(蒸馏水、醋酸铵和盐酸)对AAEMs的选择性脱除及其生物油组分的影响,研究了碱和碱土金属类别(K、Ca、Na和Mg)、盐浓度(0.5%、2.5%、5%)和不同钾盐的酸根(${\rm{SO}}_{4}^{{2-}} $ 、${\rm{NO}}_{3}^{-} $ 、${\rm{CO}}_{3}^{{2-}} $ 、${\rm{HCO}}_{3}^{-} $ 、AC−和${\rm{PO}}_{4}^{{{3-}}}$ )对生物油组分的影响。结果表明,在梯级脱灰预处理过程中,随着脱灰溶液酸性程度加深,AAEMs的脱除率逐渐上升,根据AAEMs在梯级脱灰过程中的选择性脱除规律,可将其在生物质中的赋存形态分为水溶性(K)、离子交换性(Ca和Mg)和酸溶性(Na)等形态。经过碱和碱土金属盐浸渍后,AAEMs将起到催化剂的作用,促进热解中间产物左旋葡聚糖的二次降解,导致其相对含量显著降低,形成更多的呋喃和酮类等轻质含氧化合物,导致2, 3-二氢苯并呋喃、酮类和长链烷烃等组分的含量显著增加。不同钾盐酸根离子对脱水糖的二次裂解反应及木质素芳基醚键和酚羟基的裂解反应具有较大的影响,根据酸根的酸性强弱,对脱水糖裂解反应的影响大小顺序为${\rm{HCO}}_{3}^{-} $ >${\rm{CO}}_{3}^{{{2-}}}$ >AC−>${\rm{PO}}_{4}^{{{3-}}}$ >Cl−>${\rm{NO}}_{3}^{-} $ >${\rm{SO}}_{4}^{{{2-}}}$ ,而对木质素芳基醚键和酚羟基的裂解反应影响大小顺序为${\rm{CO}}_{3}^{{{2-}}}$ >Cl−>${\rm{HCO}}_{3}^{-} $ >${\rm{PO}}_{4}^{{{3-}}}$ ≈AC−>${\rm{SO}}_{4}^{{{2-}}}$ ≈${\rm{NO}}_{3}^{-} $ 。
生物质灰分中的碱和碱土金属(AAEMs)对快速热解生物油的产率和组分分布具有显著影响。本研究选取玉米秸秆为原料,研究梯级脱灰预处理(蒸馏水、醋酸铵和盐酸)对AAEMs的选择性脱除及其生物油组分的影响,研究了碱和碱土金属类别(K、Ca、Na和Mg)、盐浓度(0.5%、2.5%、5%)和不同钾盐的酸根(











当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024011
摘要:
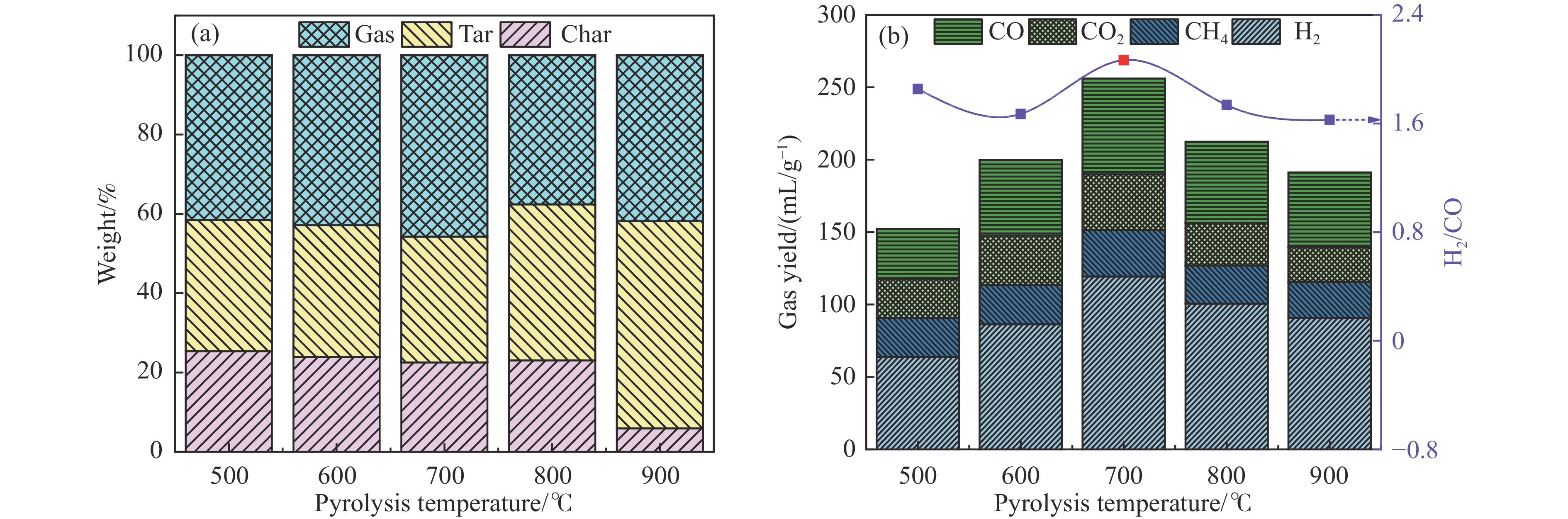
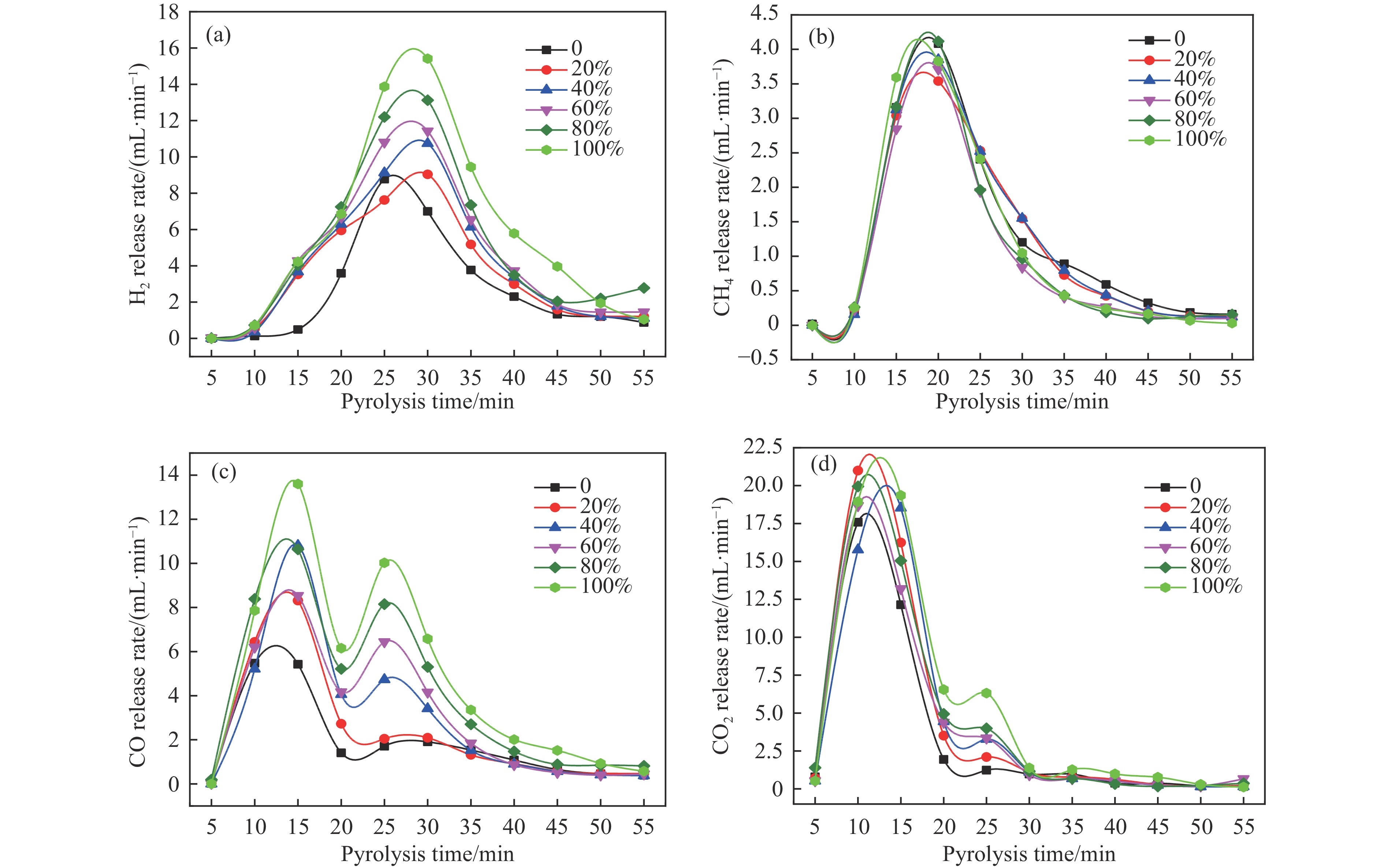
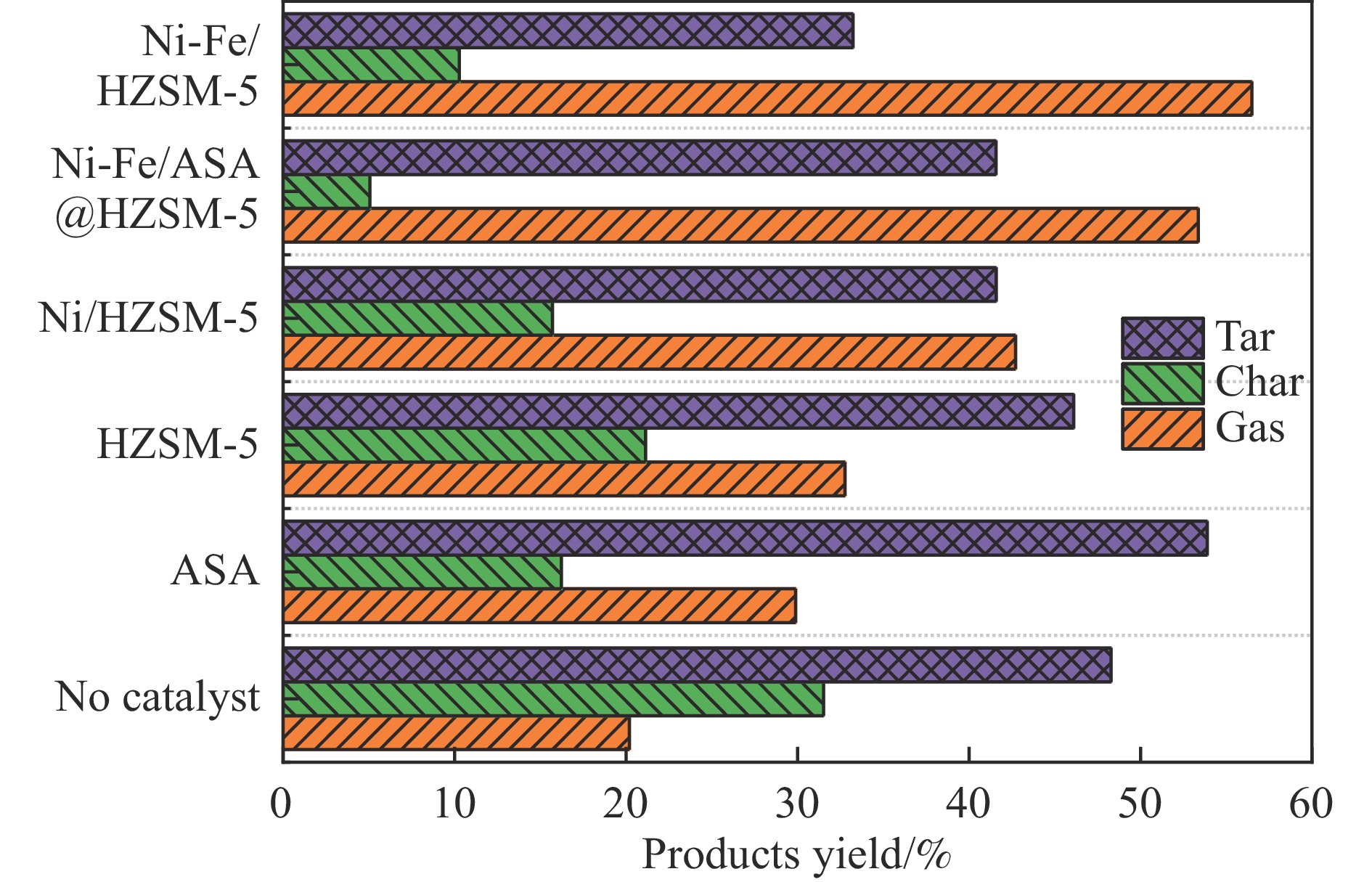
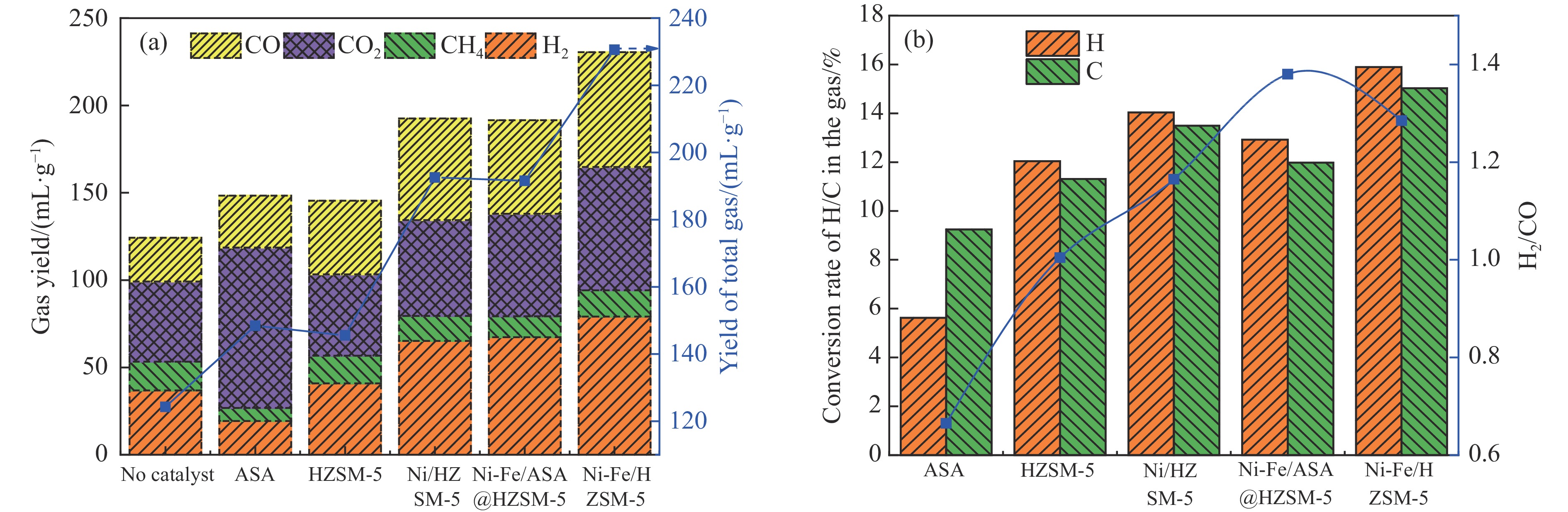
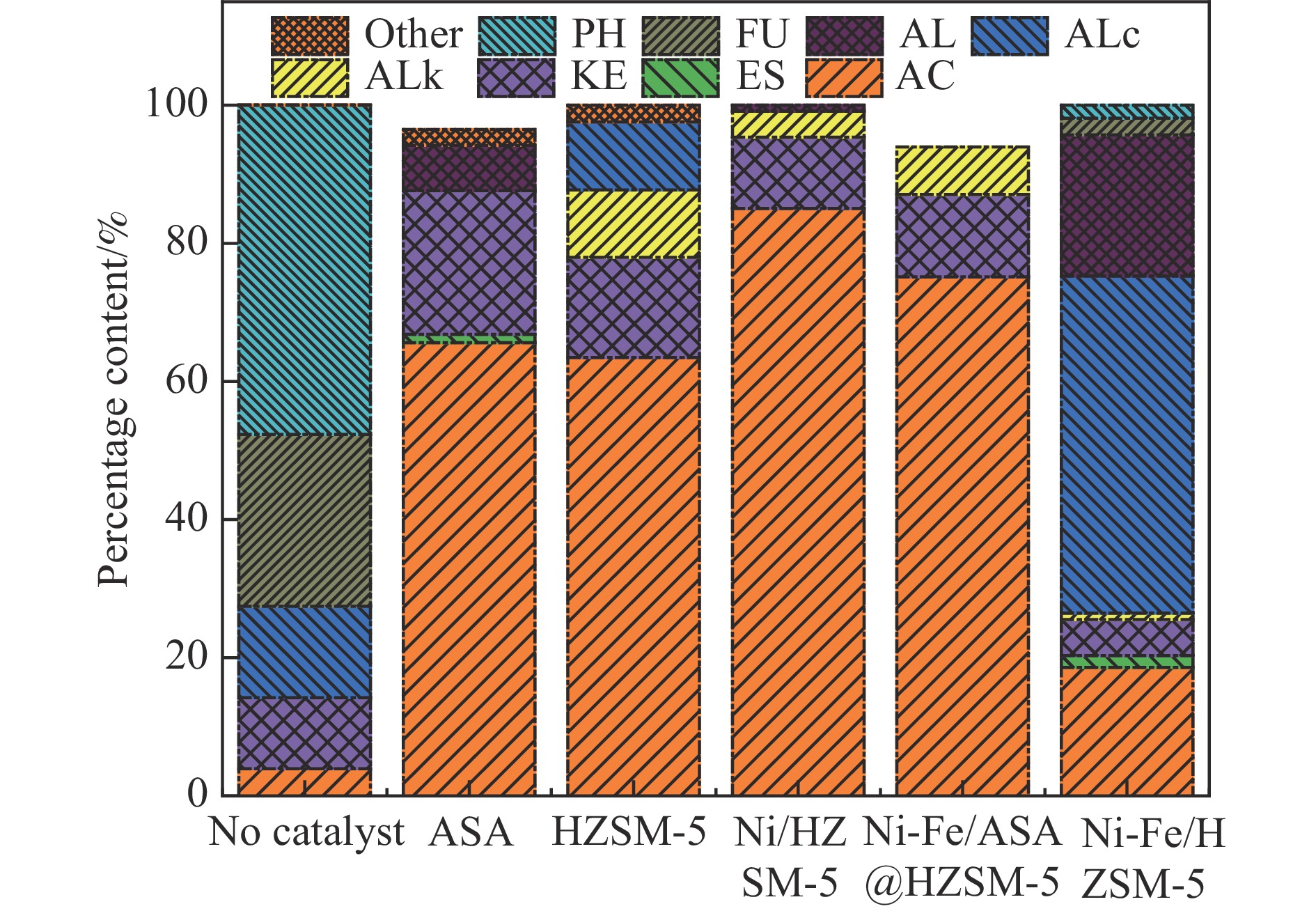
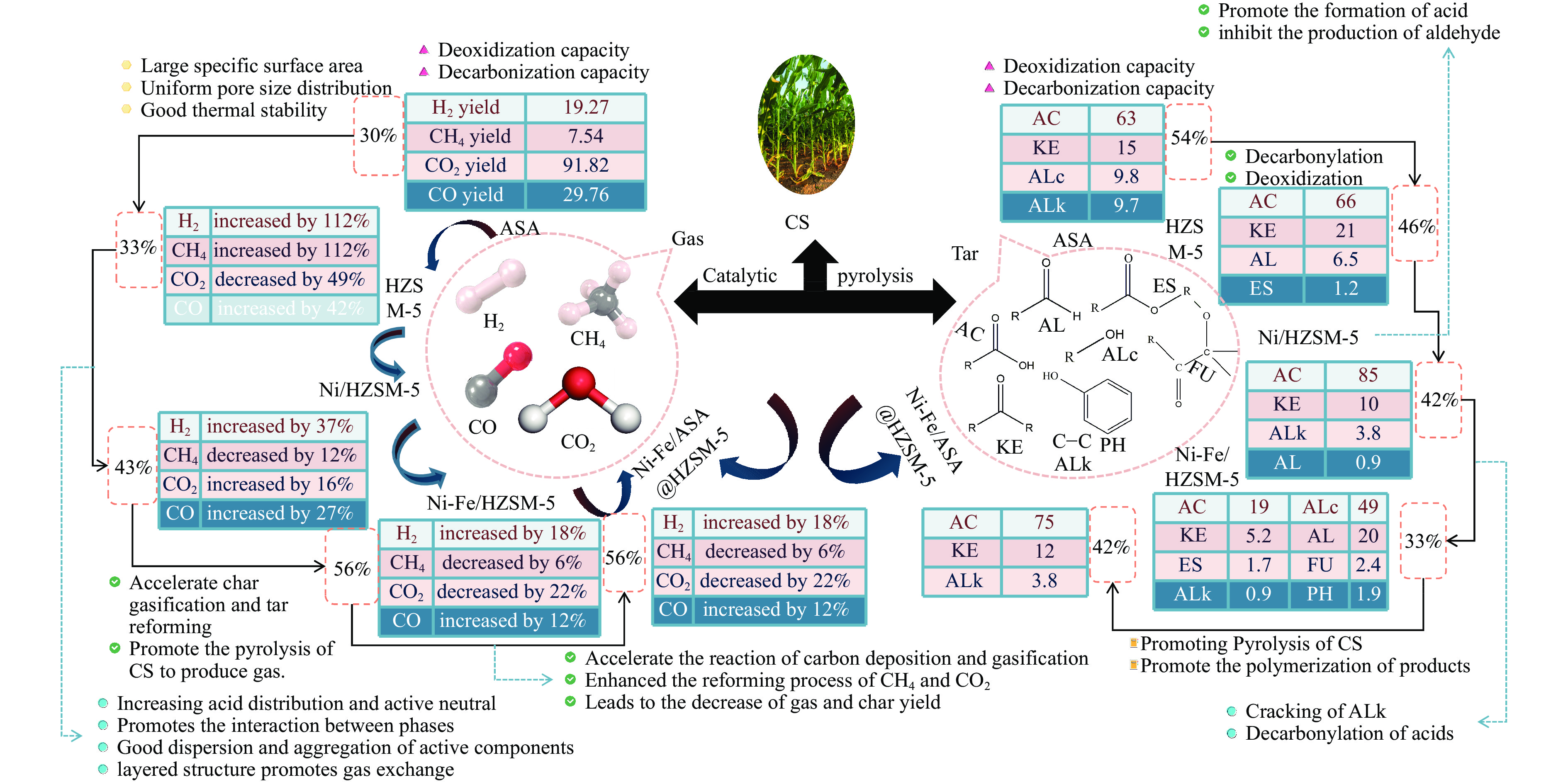
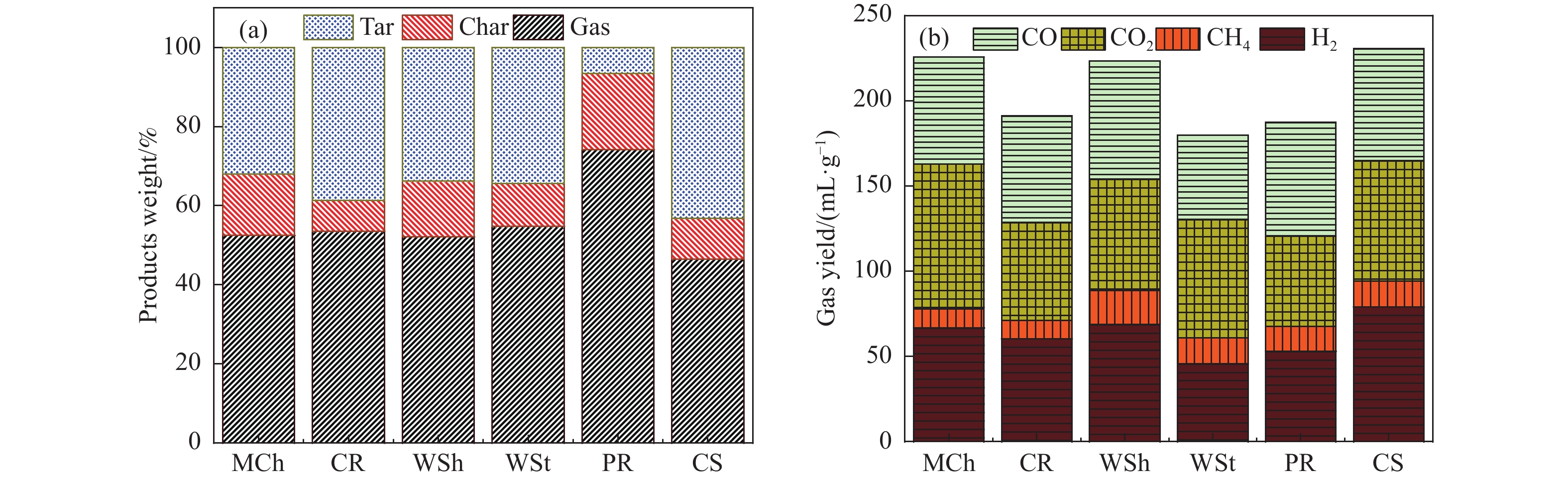
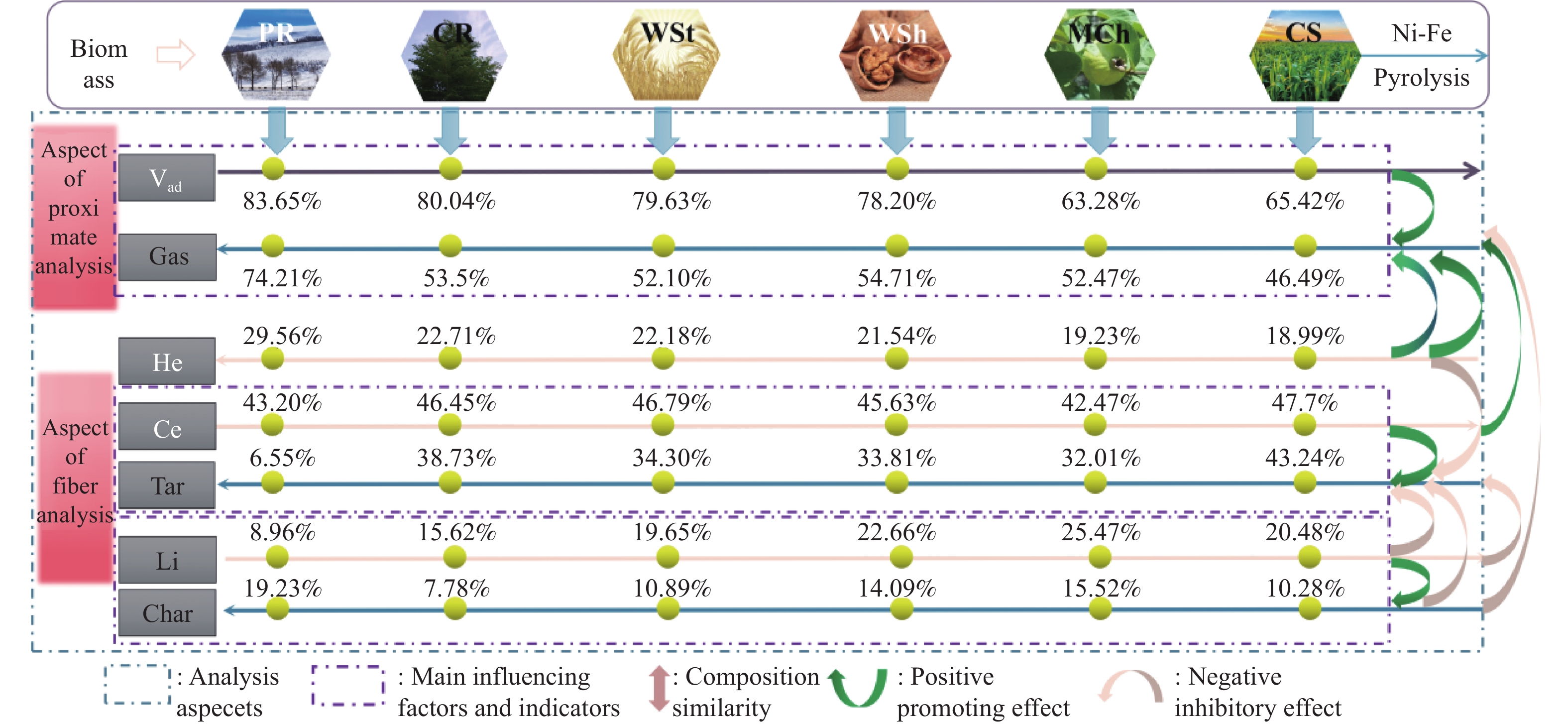
本研究通过超声波辅助过量浸渍法将活性组分镍、助剂铁与HZSM-5分子筛结合来提高富氢燃气的产率;进一步以废弃铝灰与HZSM-5分子筛作为共载体制备铝灰与HZSM-5分子筛符合共载镍-铁催化剂并将其用于强化生物质催化热解产富氢燃气的过程。结果表明,在热解温度700 ℃下,Ni-Fe/HZSM-5可使富氢燃气的产率提高到56.49%(约为230.59 mL/g),氢气产率提高到63.12%,产氢效率提高到0.71%,CO得率增加到65.77 mL/g;足够的Ni-Fe/HZSM-5催化剂量强化了生物质热解的产氢路径,促进了积炭气化反应,起到提高H2和CO产率的双重作用。不同种类生物质的组成差异导致催化热解的产物分布也不同,Ni-Fe/HZSM-5催化生物质热解气体产率的顺序为PR(74.21%)>WSt(54.71%)>CR(53.5%)>MCh(52.47%)>WSh(52.10%)>CS(46.49%)。HZSM-5和ASA载体间的协同作用强化了CH4与CO2的重整过程,抑制了逆水汽变换反应,获得了53.37%和41.56%的气体和焦油产率;并加速了积炭气化反应从而减少了积炭量(0.05g/g),获得了5.07%的半焦产率;Ni-Fe/ASA@HZSM-5具有较好的热裂化能力和脱氧能力,有助于促进HZSM-5催化剂上富氢燃气的生成;为开发高温热解气深度净化与高效利用技术提供理论支撑,有效指导多级催化重整的新型双催化床层的开发。
本研究通过超声波辅助过量浸渍法将活性组分镍、助剂铁与HZSM-5分子筛结合来提高富氢燃气的产率;进一步以废弃铝灰与HZSM-5分子筛作为共载体制备铝灰与HZSM-5分子筛符合共载镍-铁催化剂并将其用于强化生物质催化热解产富氢燃气的过程。结果表明,在热解温度700 ℃下,Ni-Fe/HZSM-5可使富氢燃气的产率提高到56.49%(约为230.59 mL/g),氢气产率提高到63.12%,产氢效率提高到0.71%,CO得率增加到65.77 mL/g;足够的Ni-Fe/HZSM-5催化剂量强化了生物质热解的产氢路径,促进了积炭气化反应,起到提高H2和CO产率的双重作用。不同种类生物质的组成差异导致催化热解的产物分布也不同,Ni-Fe/HZSM-5催化生物质热解气体产率的顺序为PR(74.21%)>WSt(54.71%)>CR(53.5%)>MCh(52.47%)>WSh(52.10%)>CS(46.49%)。HZSM-5和ASA载体间的协同作用强化了CH4与CO2的重整过程,抑制了逆水汽变换反应,获得了53.37%和41.56%的气体和焦油产率;并加速了积炭气化反应从而减少了积炭量(0.05g/g),获得了5.07%的半焦产率;Ni-Fe/ASA@HZSM-5具有较好的热裂化能力和脱氧能力,有助于促进HZSM-5催化剂上富氢燃气的生成;为开发高温热解气深度净化与高效利用技术提供理论支撑,有效指导多级催化重整的新型双催化床层的开发。


当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60441-X
摘要:
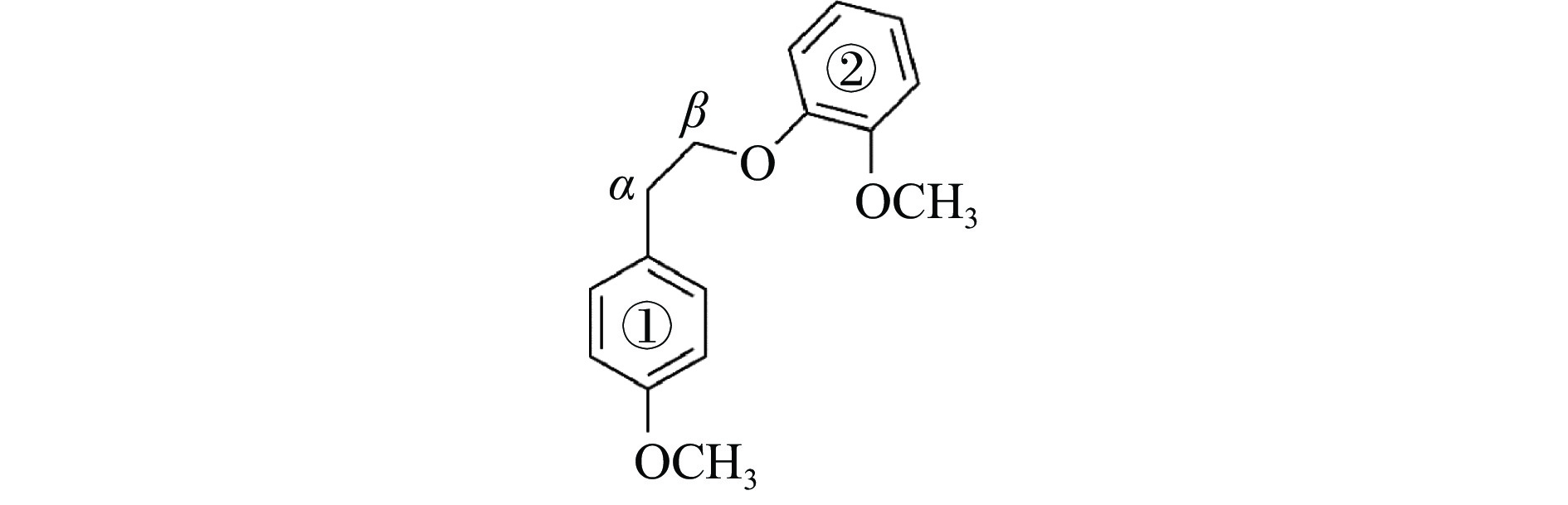
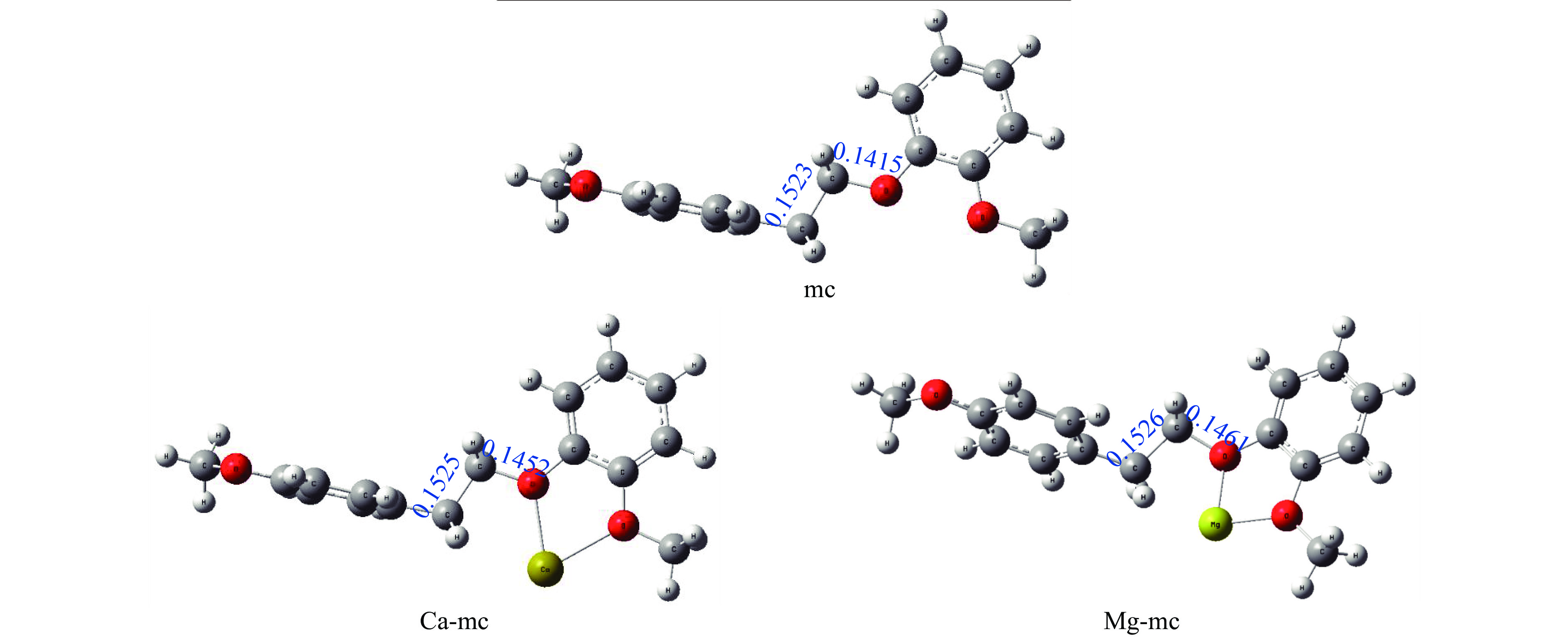
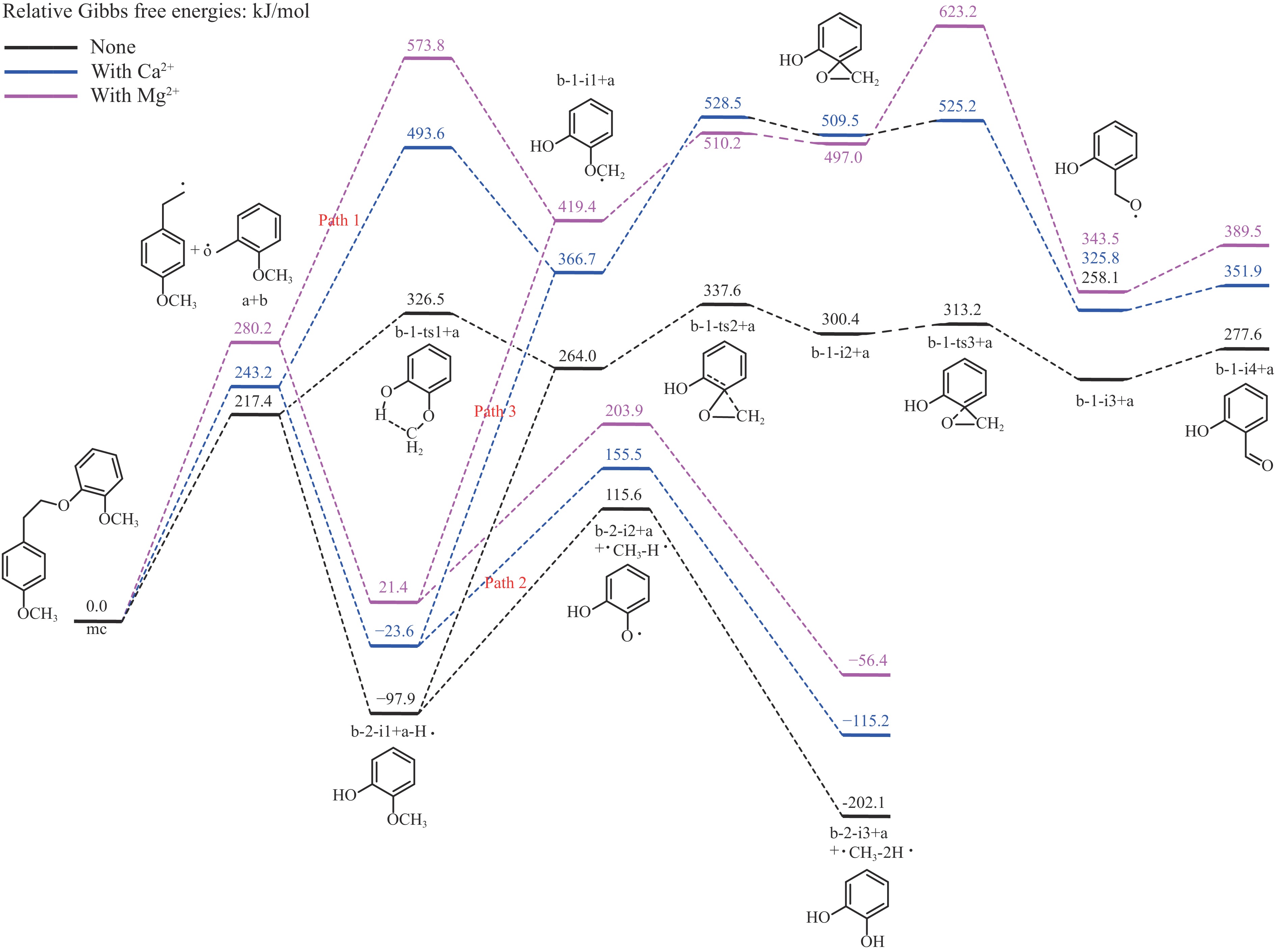
It is essential to investigate the influence of alkaline earth metals on the pyrolysis mechanism and resulting products of lignin to enhance the efficient thermochemical conversion and utilization of lignin or biomass. In this study, the Density functional theory method was used to simulate the pyrolytic reaction pathways of a β–O–4 type lignin dimer model compound (1-methoxy-2-(4-methoxyphenethoxy)benzene, mc) affected by alkaline earth metal ions Ca2+ and Mg2+. The computational findings suggest that Ca2+ and Mg2+ tend to combine with the oxygen atom at the Cβ position and the oxygen atom on the methoxy group of the lignin dimer model compound, forming stable complexes that modify the bond lengths of the Cα–Cβ and Cβ–O bonds and affect their pyrolysis energy barriers. During the catalytic pyrolysis process, the presence of Ca2+ and Mg2+ can promote the concerted decomposition reaction, leading to increased production of products like 1-methoxy-4-vinylbenzene, 2-methoxyphenol and catechol. Meanwhile, they can suppress homolytic cleavage reactions of the Cβ–O and Cα–Cβ bonds, thereby hindering the formation of other products such as 1-ethyl-4-methoxybenzene and 2-hydroxybenzaldehyde.
It is essential to investigate the influence of alkaline earth metals on the pyrolysis mechanism and resulting products of lignin to enhance the efficient thermochemical conversion and utilization of lignin or biomass. In this study, the Density functional theory method was used to simulate the pyrolytic reaction pathways of a β–O–4 type lignin dimer model compound (1-methoxy-2-(4-methoxyphenethoxy)benzene, mc) affected by alkaline earth metal ions Ca2+ and Mg2+. The computational findings suggest that Ca2+ and Mg2+ tend to combine with the oxygen atom at the Cβ position and the oxygen atom on the methoxy group of the lignin dimer model compound, forming stable complexes that modify the bond lengths of the Cα–Cβ and Cβ–O bonds and affect their pyrolysis energy barriers. During the catalytic pyrolysis process, the presence of Ca2+ and Mg2+ can promote the concerted decomposition reaction, leading to increased production of products like 1-methoxy-4-vinylbenzene, 2-methoxyphenol and catechol. Meanwhile, they can suppress homolytic cleavage reactions of the Cβ–O and Cα–Cβ bonds, thereby hindering the formation of other products such as 1-ethyl-4-methoxybenzene and 2-hydroxybenzaldehyde.
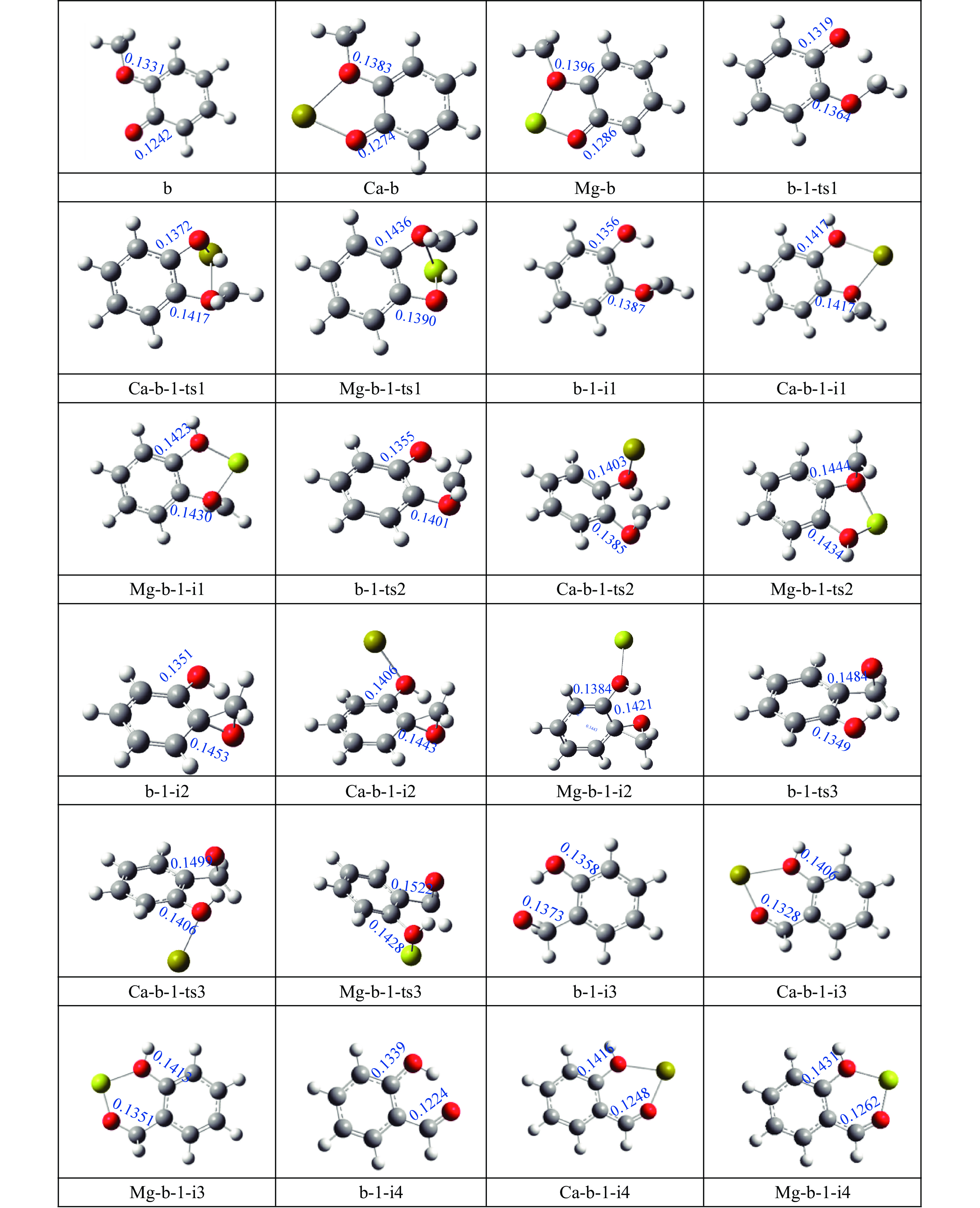
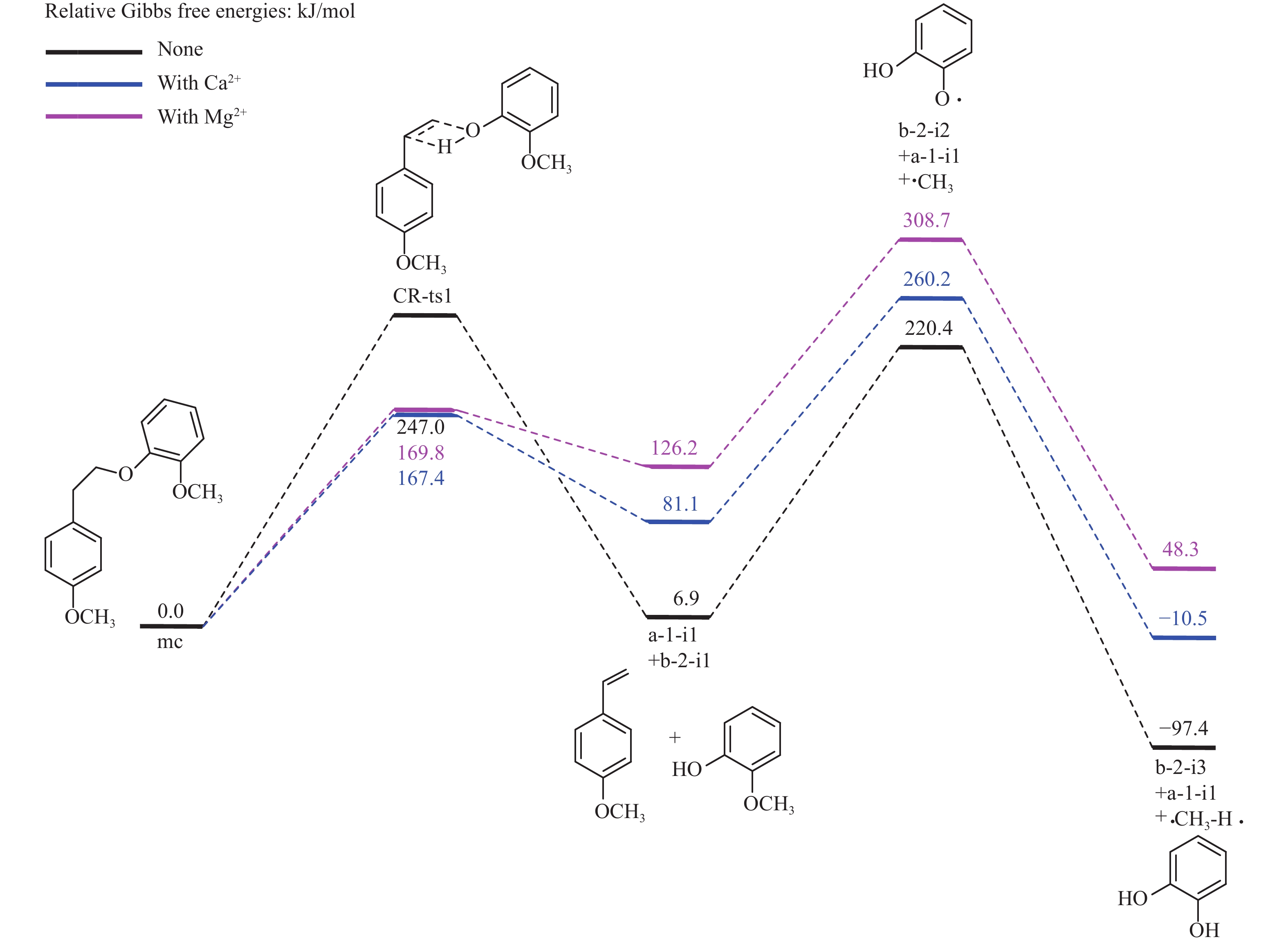
当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024009
摘要:
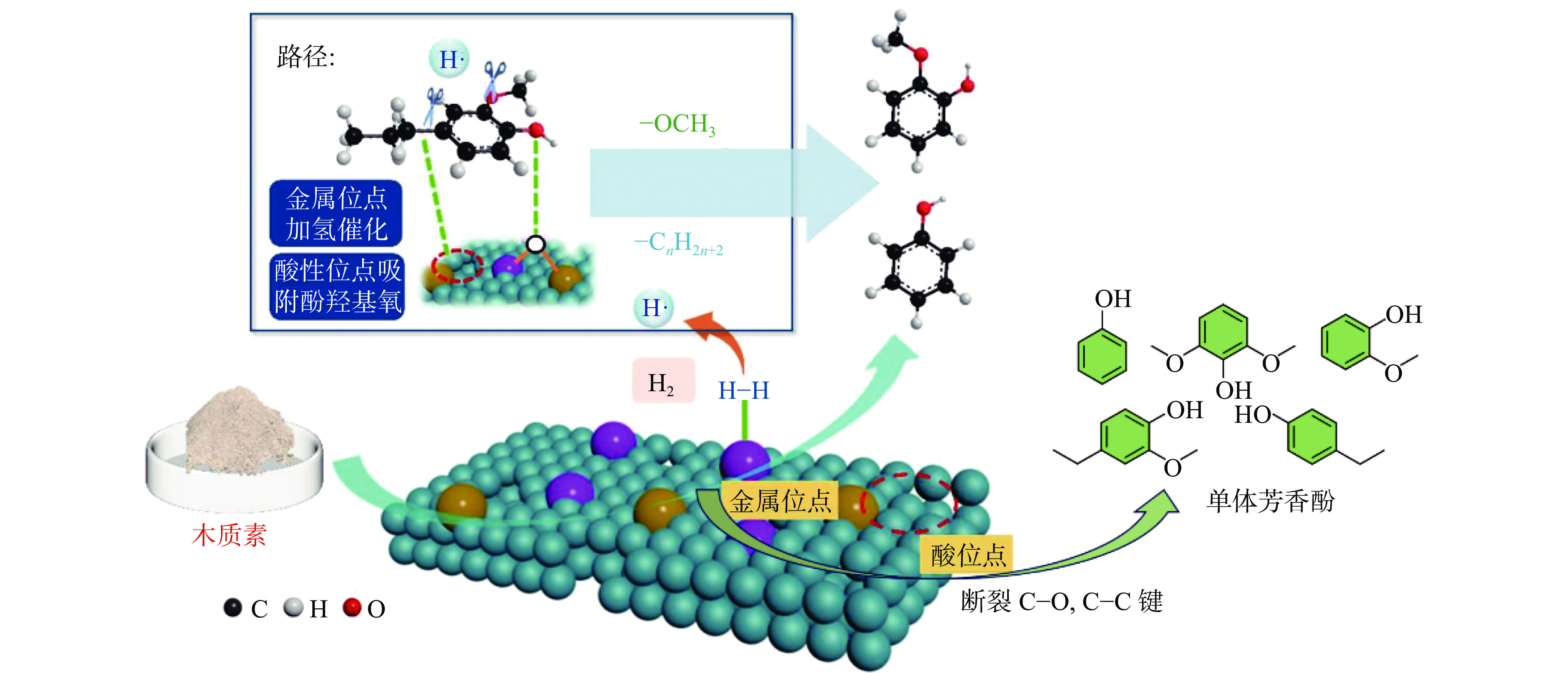
开发和利用可再生生物质资源是实现二氧化碳减排的有效途径。而生物质结构复杂,整体反应性和利用率较低。木质素是自然界中唯一具有高能量密度的可再生芳香族聚合物,其转化和利用在全球范围内引起了广泛关注。然而,木质素结构的复杂性、连接方式的不确定性、侧链连接的稳定性以及反应片段不可避免的再缩合,使得将木质素解聚成生物燃料或芳香化学品成为一项艰巨的挑战。催化氢解技术可将木质素转化为高选择性、高收率的酚类单体。但是木质素催化解聚过程中化学键的定向剪切,产物与结构之间的转化机理仍不清晰。本工作针对木质素的催化氢解生产高值化学品的最新研究进展,重点总结了木质素的催化氢解过程中催化剂及其高值化学品产物间的耦合关联,着重讨论了不同催化剂体系对木质素解聚产物过程机理的影响,尤其对于金属基催化剂,综述了贵金属基催化剂、过渡基金属催化剂、水滑石催化剂和金属有机框架催化剂对于产物分布影响的最新进展,并进一步总结了不同催化剂存在的问题和转化机制;同时木质素加氢催化裂解过程中溶剂是促进木质素溶解、加速传热传质、促进反应物和催化剂在反应器中均匀分散的关键。本工作并对木质素液化的主要溶剂,例如水、醇类和新型溶剂体系对木质素的解聚效应进行综述。最后,就领域所面临的机遇和挑战进行了总结和展望,为木质素高效定向转化与高值化利用提供了理论参考。
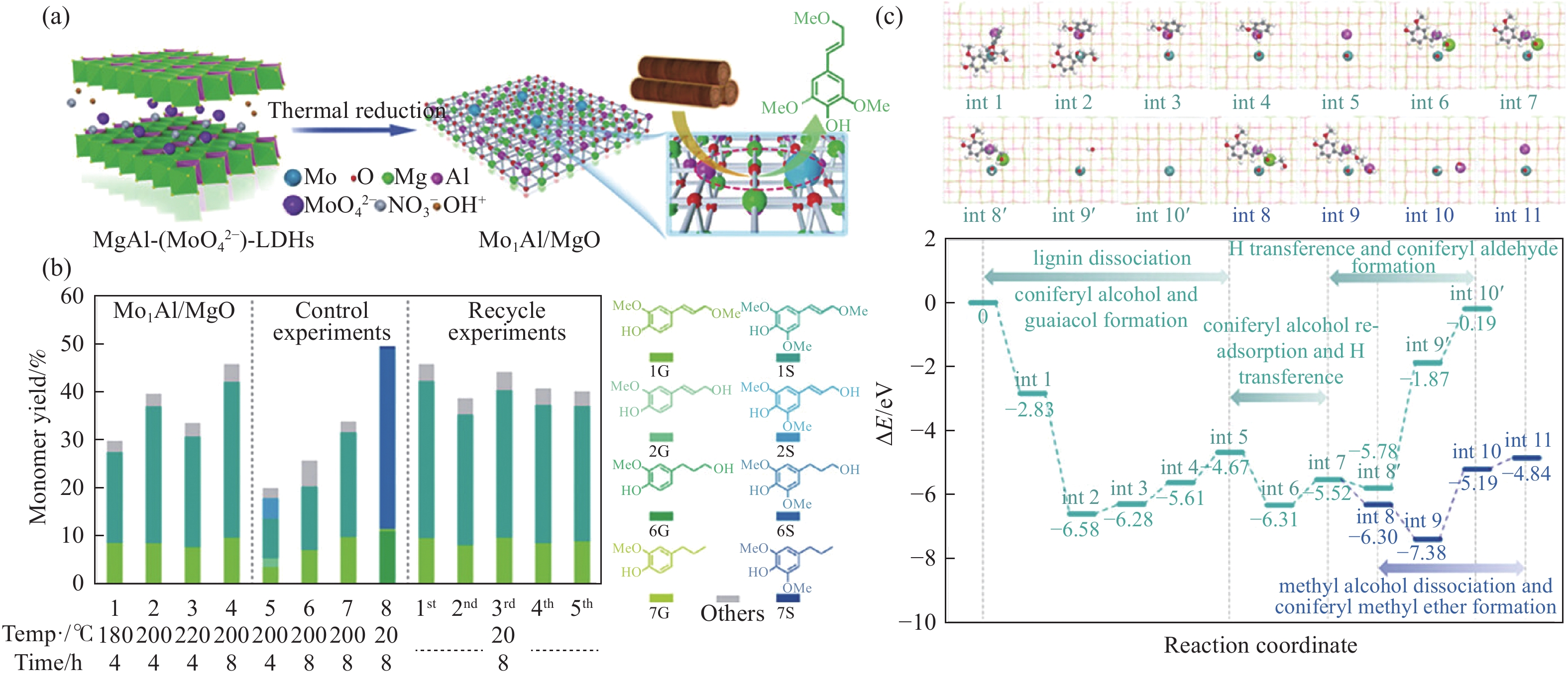
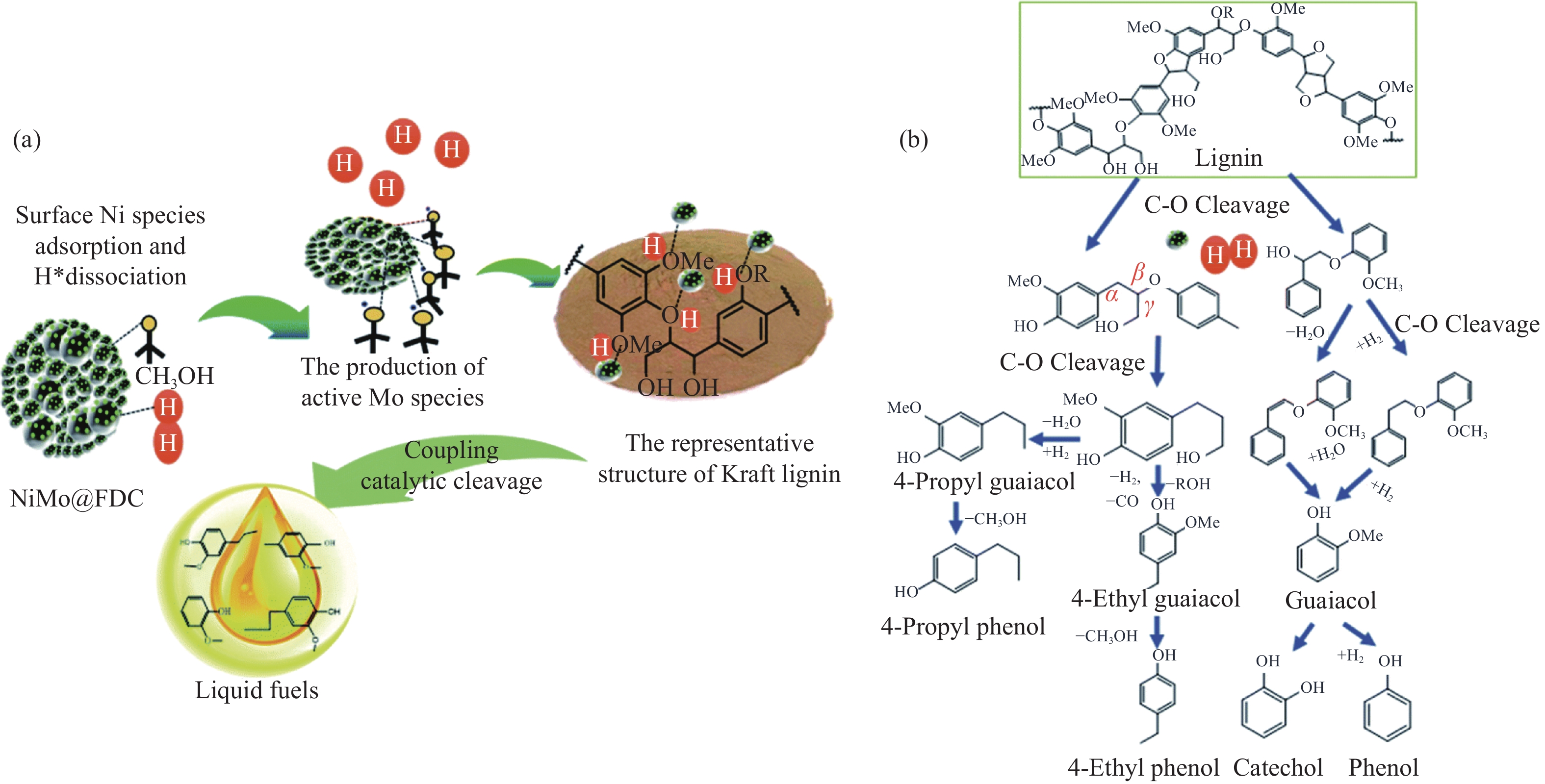
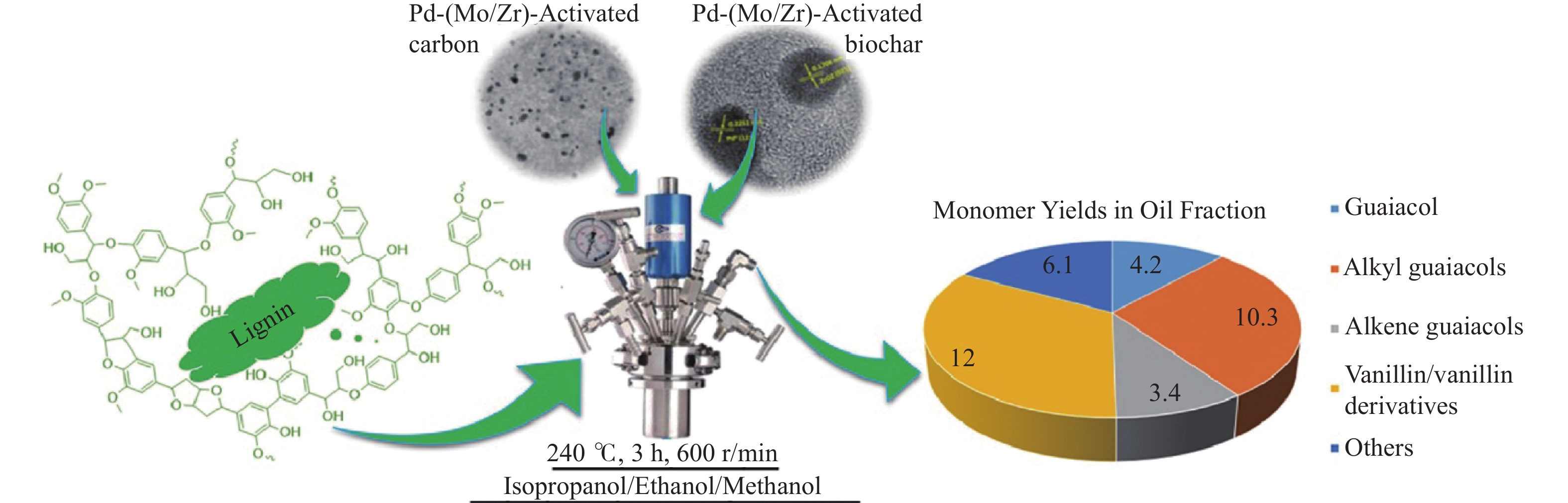
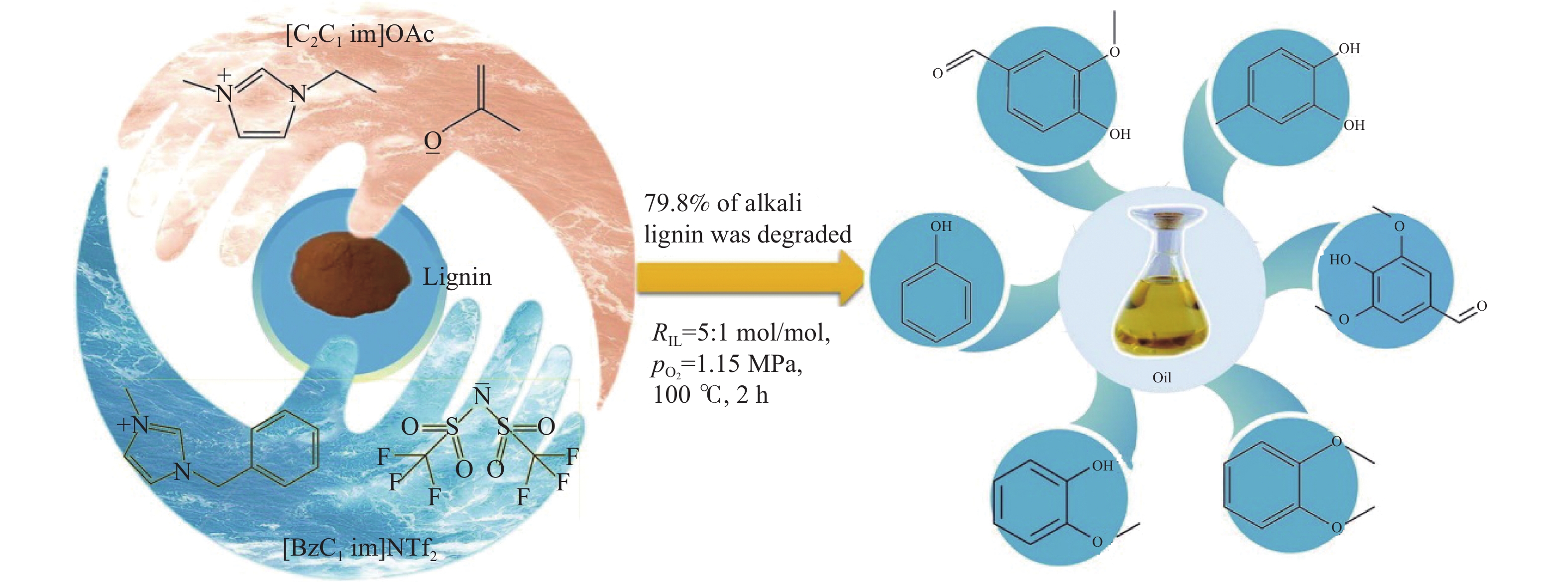
开发和利用可再生生物质资源是实现二氧化碳减排的有效途径。而生物质结构复杂,整体反应性和利用率较低。木质素是自然界中唯一具有高能量密度的可再生芳香族聚合物,其转化和利用在全球范围内引起了广泛关注。然而,木质素结构的复杂性、连接方式的不确定性、侧链连接的稳定性以及反应片段不可避免的再缩合,使得将木质素解聚成生物燃料或芳香化学品成为一项艰巨的挑战。催化氢解技术可将木质素转化为高选择性、高收率的酚类单体。但是木质素催化解聚过程中化学键的定向剪切,产物与结构之间的转化机理仍不清晰。本工作针对木质素的催化氢解生产高值化学品的最新研究进展,重点总结了木质素的催化氢解过程中催化剂及其高值化学品产物间的耦合关联,着重讨论了不同催化剂体系对木质素解聚产物过程机理的影响,尤其对于金属基催化剂,综述了贵金属基催化剂、过渡基金属催化剂、水滑石催化剂和金属有机框架催化剂对于产物分布影响的最新进展,并进一步总结了不同催化剂存在的问题和转化机制;同时木质素加氢催化裂解过程中溶剂是促进木质素溶解、加速传热传质、促进反应物和催化剂在反应器中均匀分散的关键。本工作并对木质素液化的主要溶剂,例如水、醇类和新型溶剂体系对木质素的解聚效应进行综述。最后,就领域所面临的机遇和挑战进行了总结和展望,为木质素高效定向转化与高值化利用提供了理论参考。


当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60409-8
摘要:
电厂烟气主要成分为N2、CO2和部分O2,将电厂烟气注入矿井采空区可实现CO2封存,并替代注氮气防治遗煤自燃,但是烟气中的O2是造成遗煤自燃的因素之一。因此,亟待开发一种经济有效的催化剂来脱除电厂烟气中的O2。本研究采用共沉淀法,通过调变载体和负载量可控制备了铜基催化剂和系列xCuO/CeO2催化剂,利用BET、XRD、ICP、TEM、H2-TPR和XPS等手段对催化剂进行了表征,并建立催化剂结构与催化电厂烟气脱氧性能之间的构效关系。结果表明,CeO2的加入提高了CuO的分散性、增加了催化剂的氧空位,提高了催化剂的活性和还原氧化性能,Cu-Ce界面结构的协同效应促进了还原氧化过程,表现出良好的活性和循环稳定性。30CuO/CeO2由于其CuO颗粒尺寸最小、分散性最高、氧空位浓度最高,表现出较优的催化电厂烟气脱氧性能。本研究为开发低成本可循环使用、高活性和高稳定性的脱氧催化剂提供了参考。
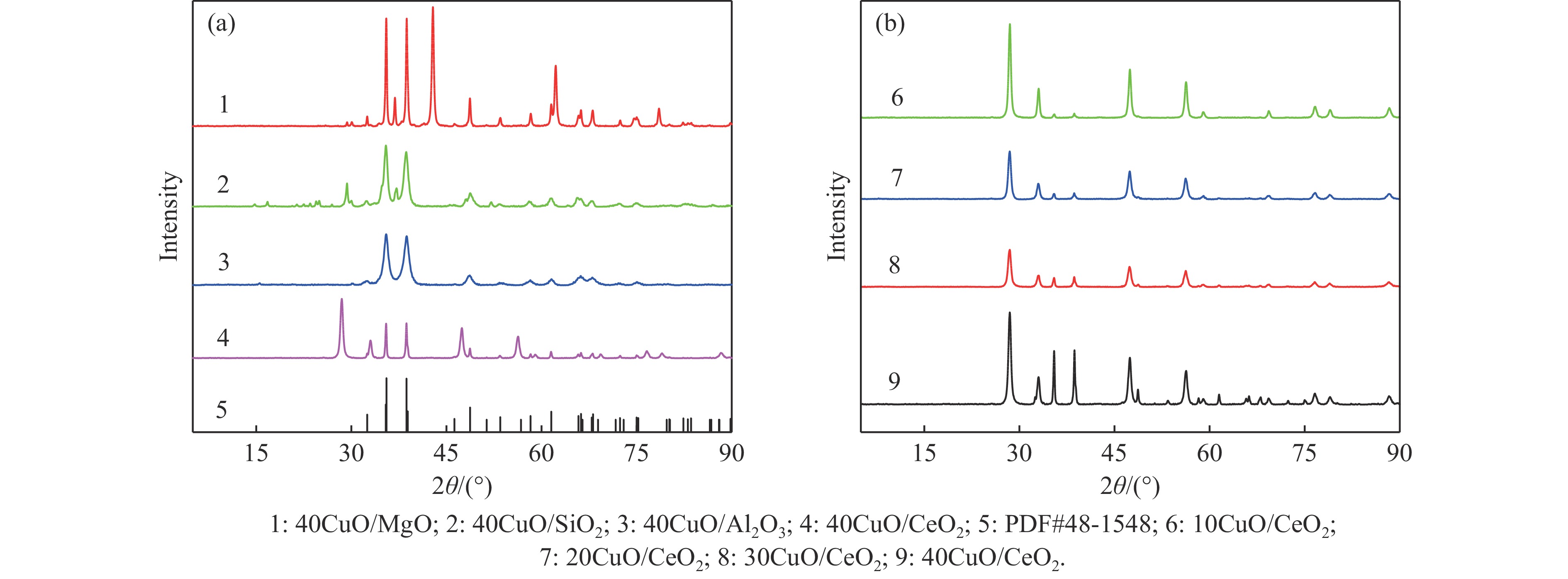
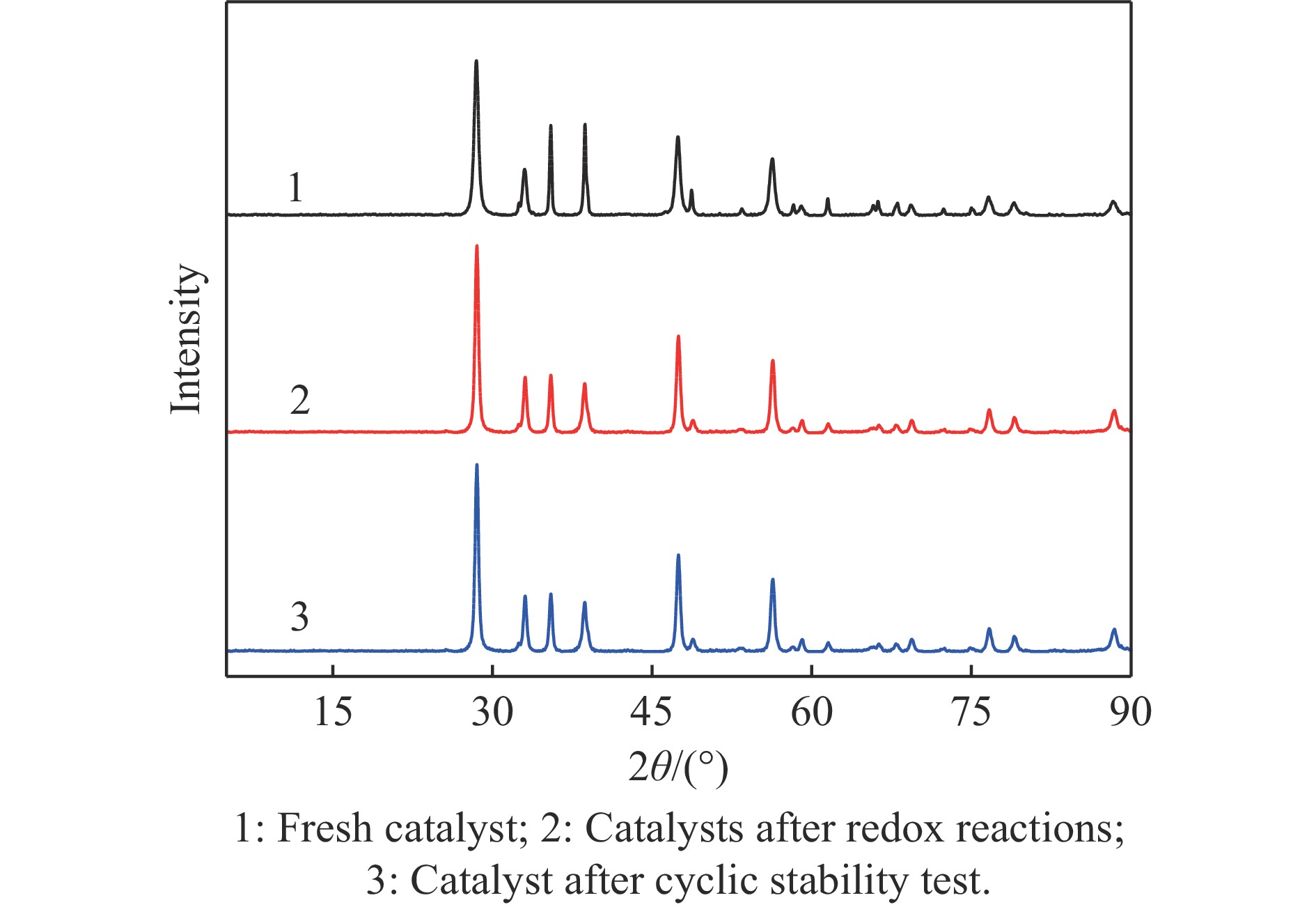
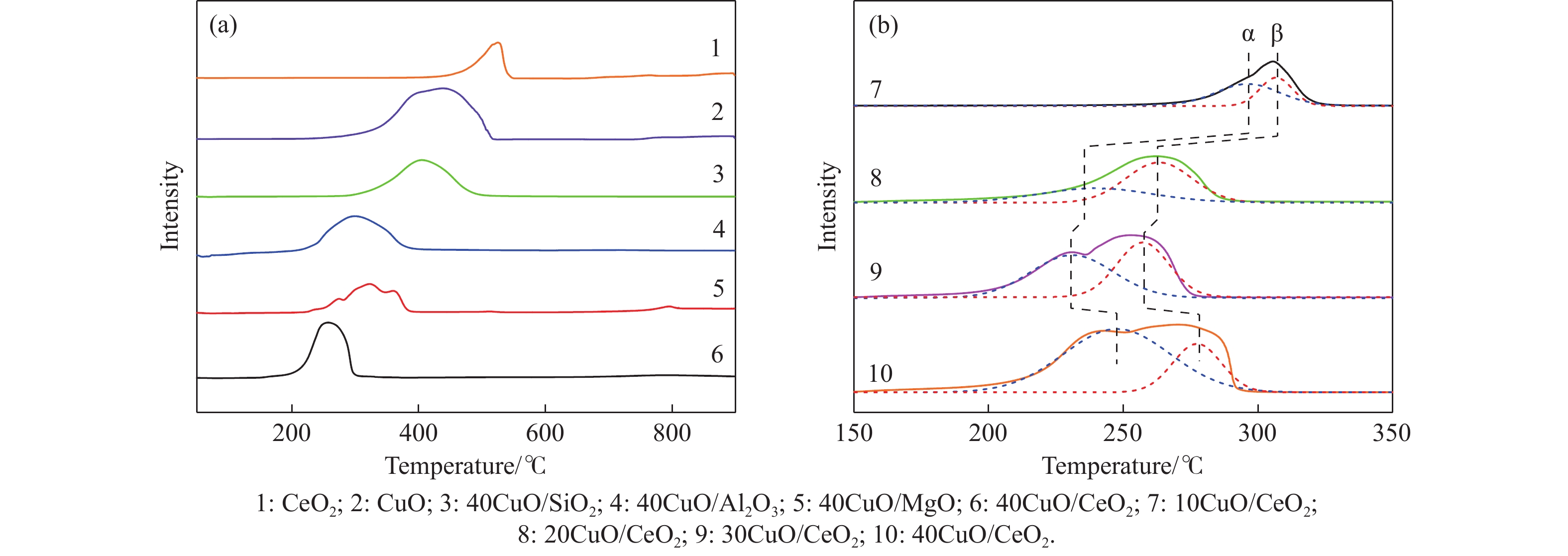
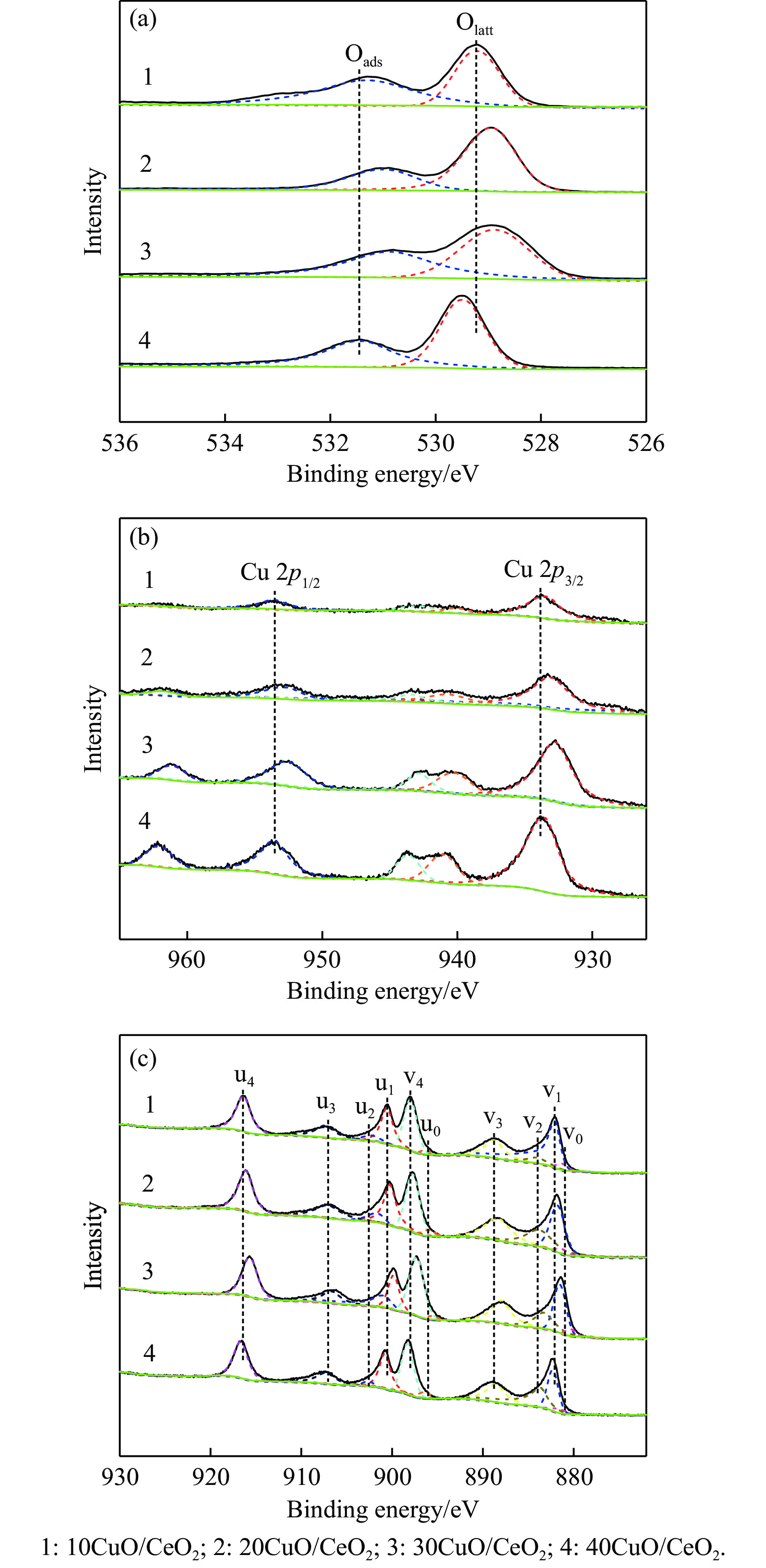
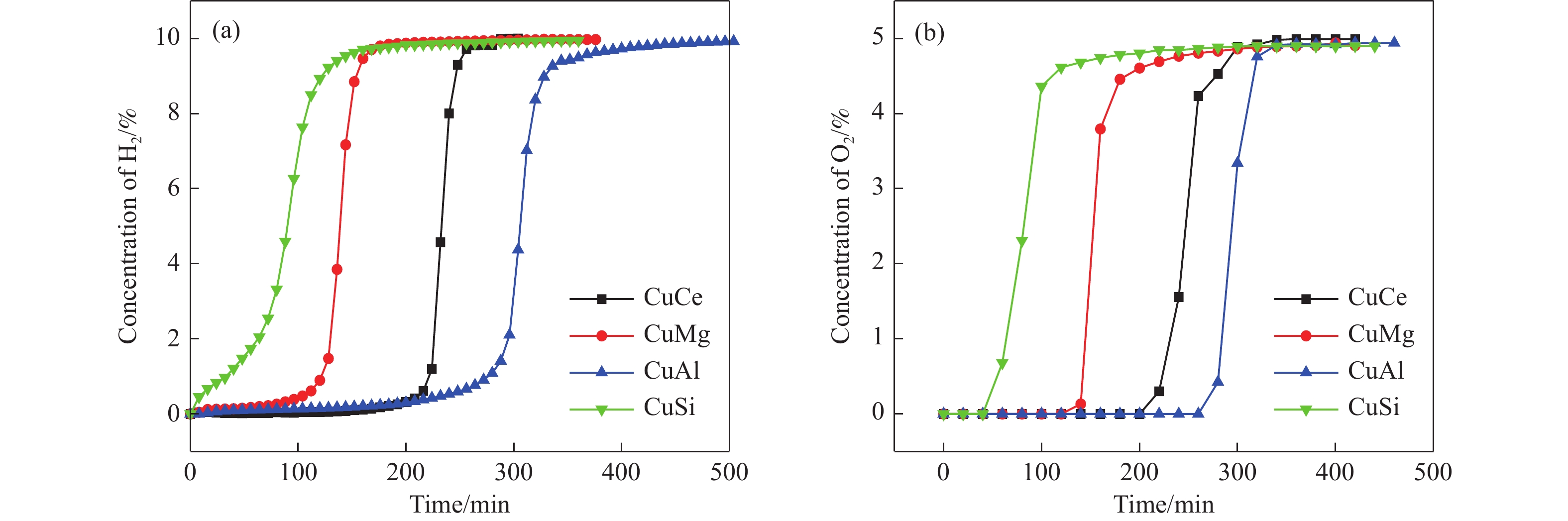
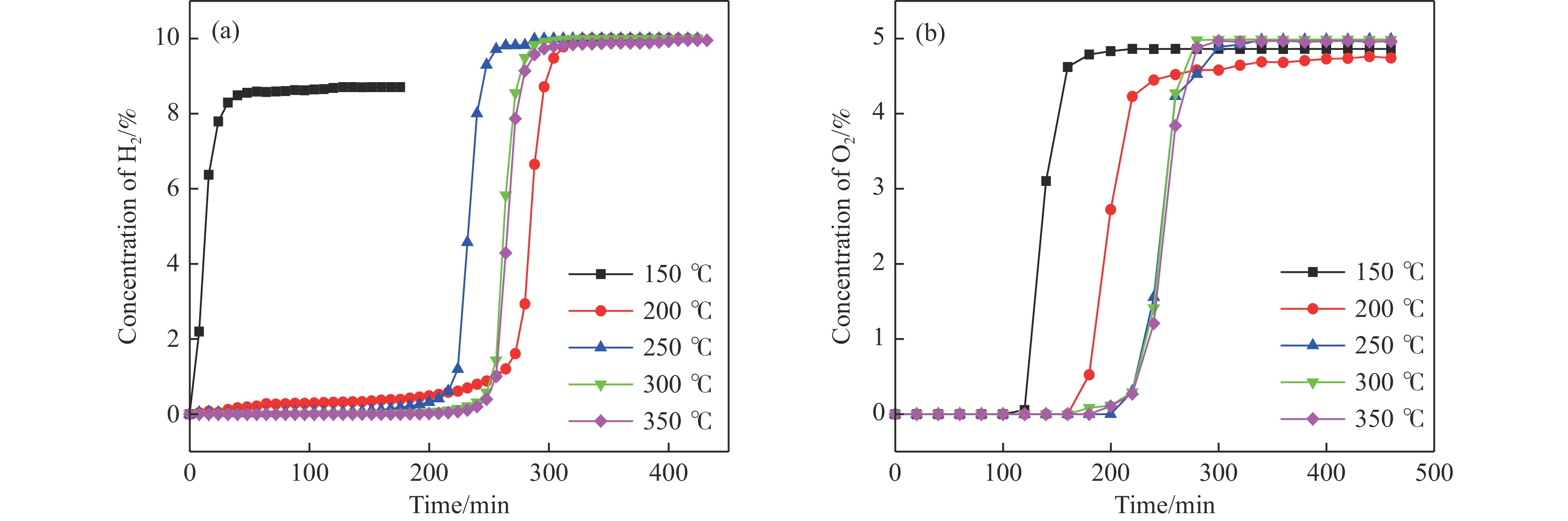
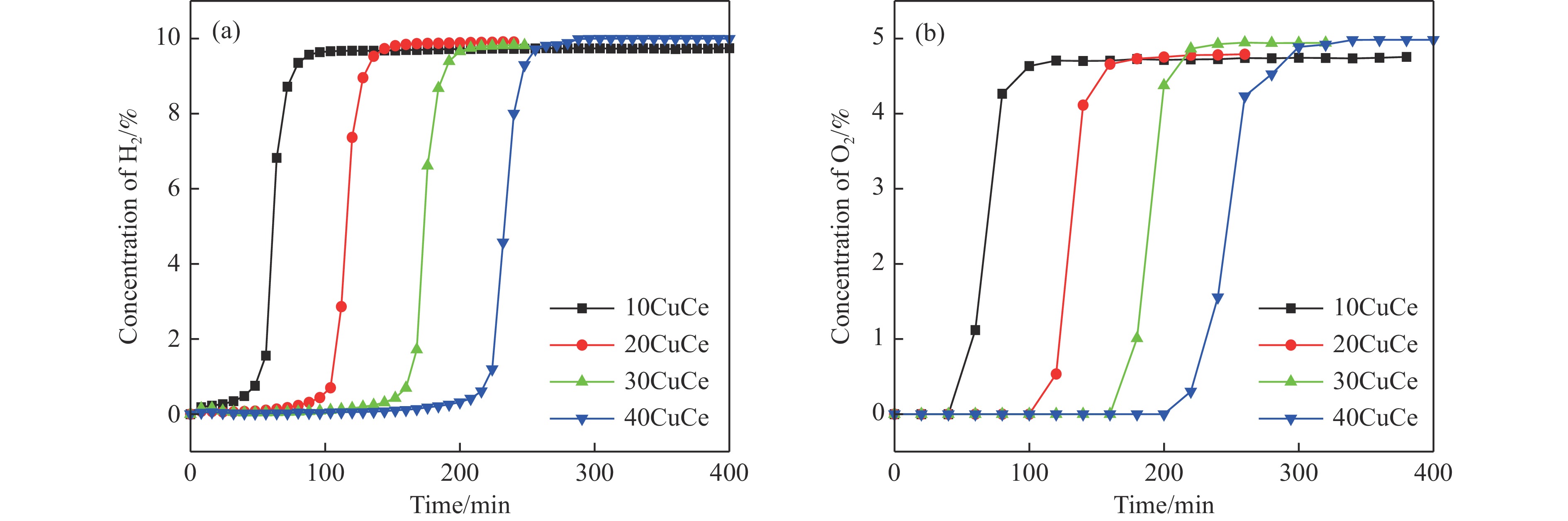
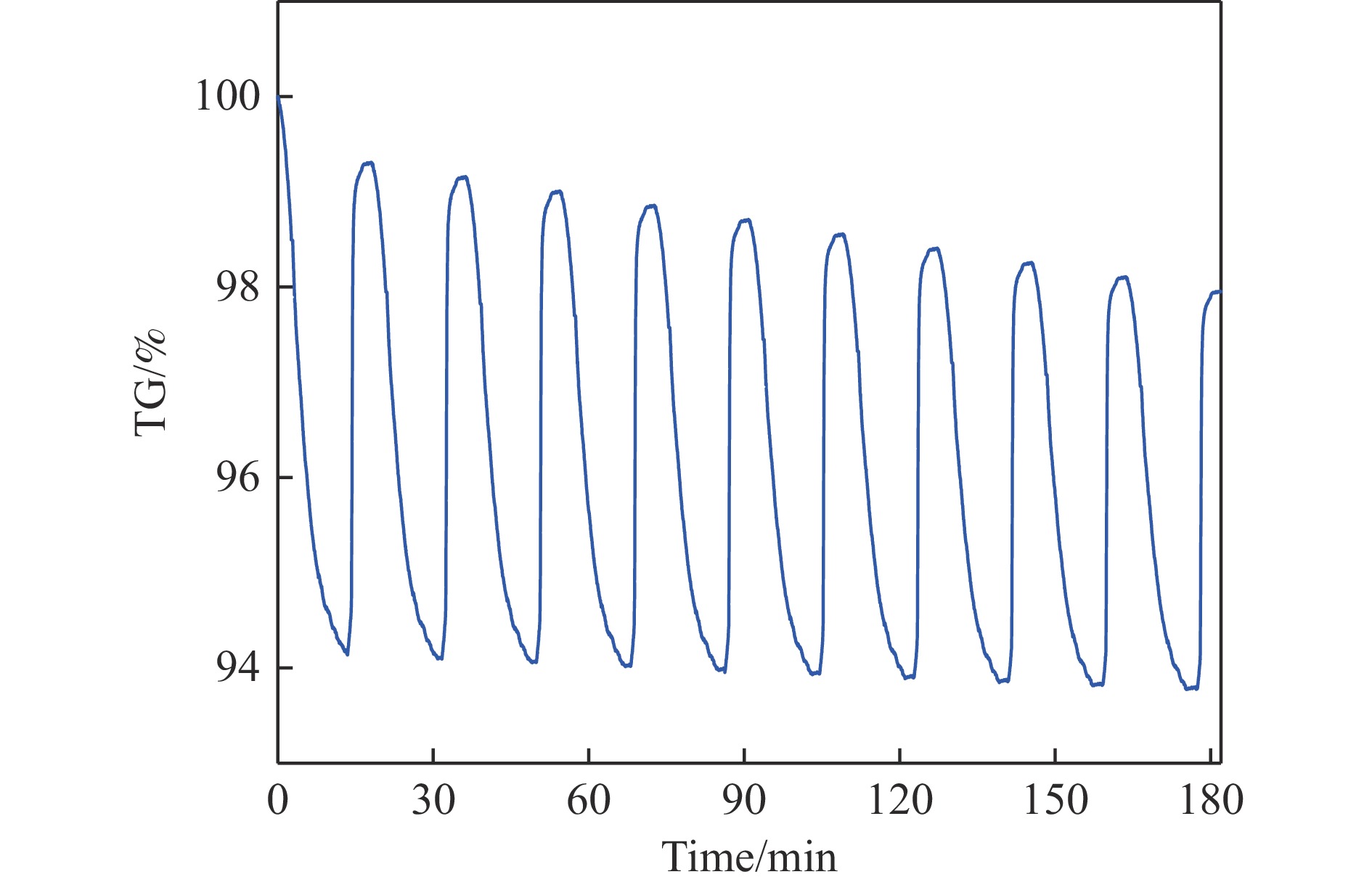
电厂烟气主要成分为N2、CO2和部分O2,将电厂烟气注入矿井采空区可实现CO2封存,并替代注氮气防治遗煤自燃,但是烟气中的O2是造成遗煤自燃的因素之一。因此,亟待开发一种经济有效的催化剂来脱除电厂烟气中的O2。本研究采用共沉淀法,通过调变载体和负载量可控制备了铜基催化剂和系列xCuO/CeO2催化剂,利用BET、XRD、ICP、TEM、H2-TPR和XPS等手段对催化剂进行了表征,并建立催化剂结构与催化电厂烟气脱氧性能之间的构效关系。结果表明,CeO2的加入提高了CuO的分散性、增加了催化剂的氧空位,提高了催化剂的活性和还原氧化性能,Cu-Ce界面结构的协同效应促进了还原氧化过程,表现出良好的活性和循环稳定性。30CuO/CeO2由于其CuO颗粒尺寸最小、分散性最高、氧空位浓度最高,表现出较优的催化电厂烟气脱氧性能。本研究为开发低成本可循环使用、高活性和高稳定性的脱氧催化剂提供了参考。




当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(24)60436-6
摘要:
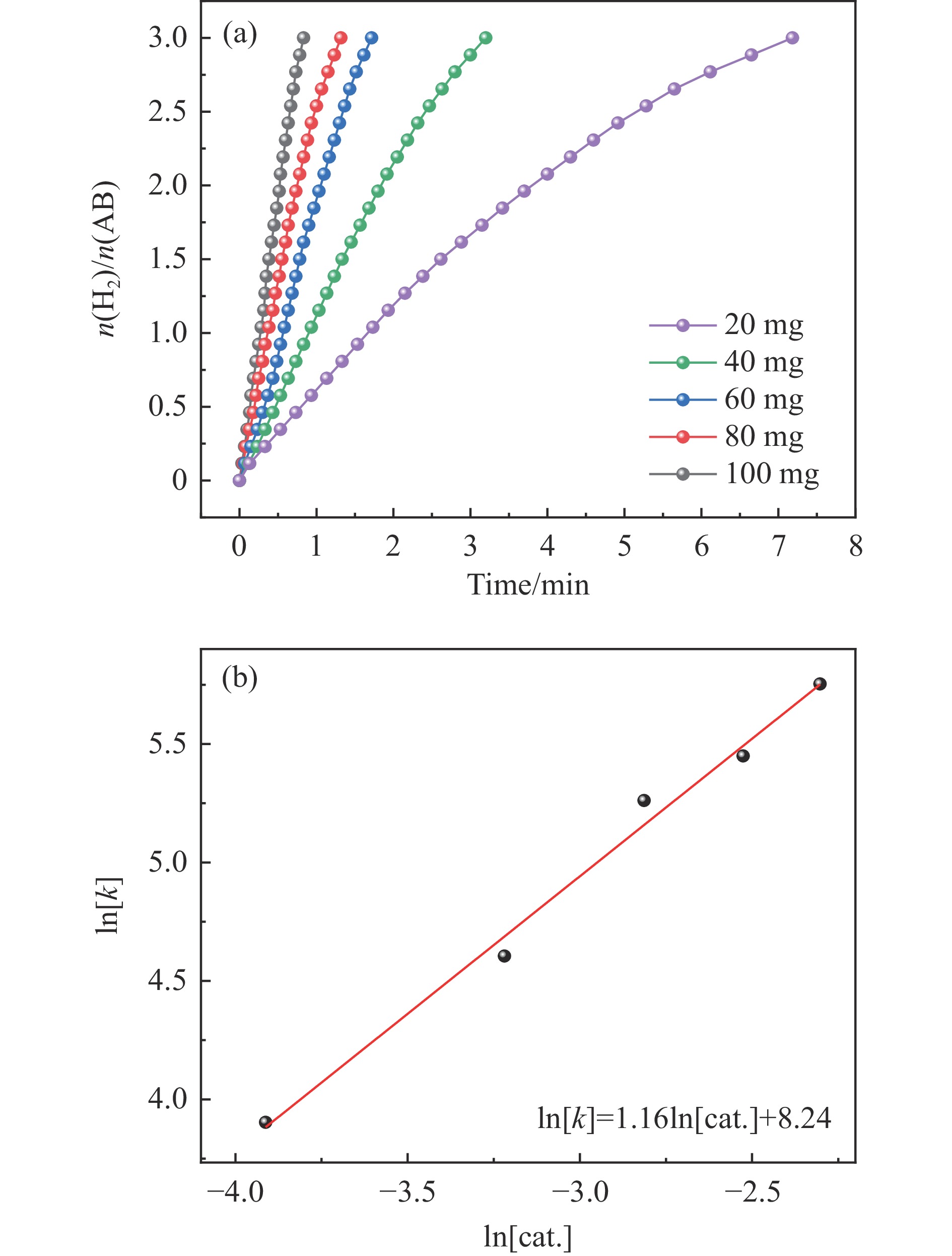
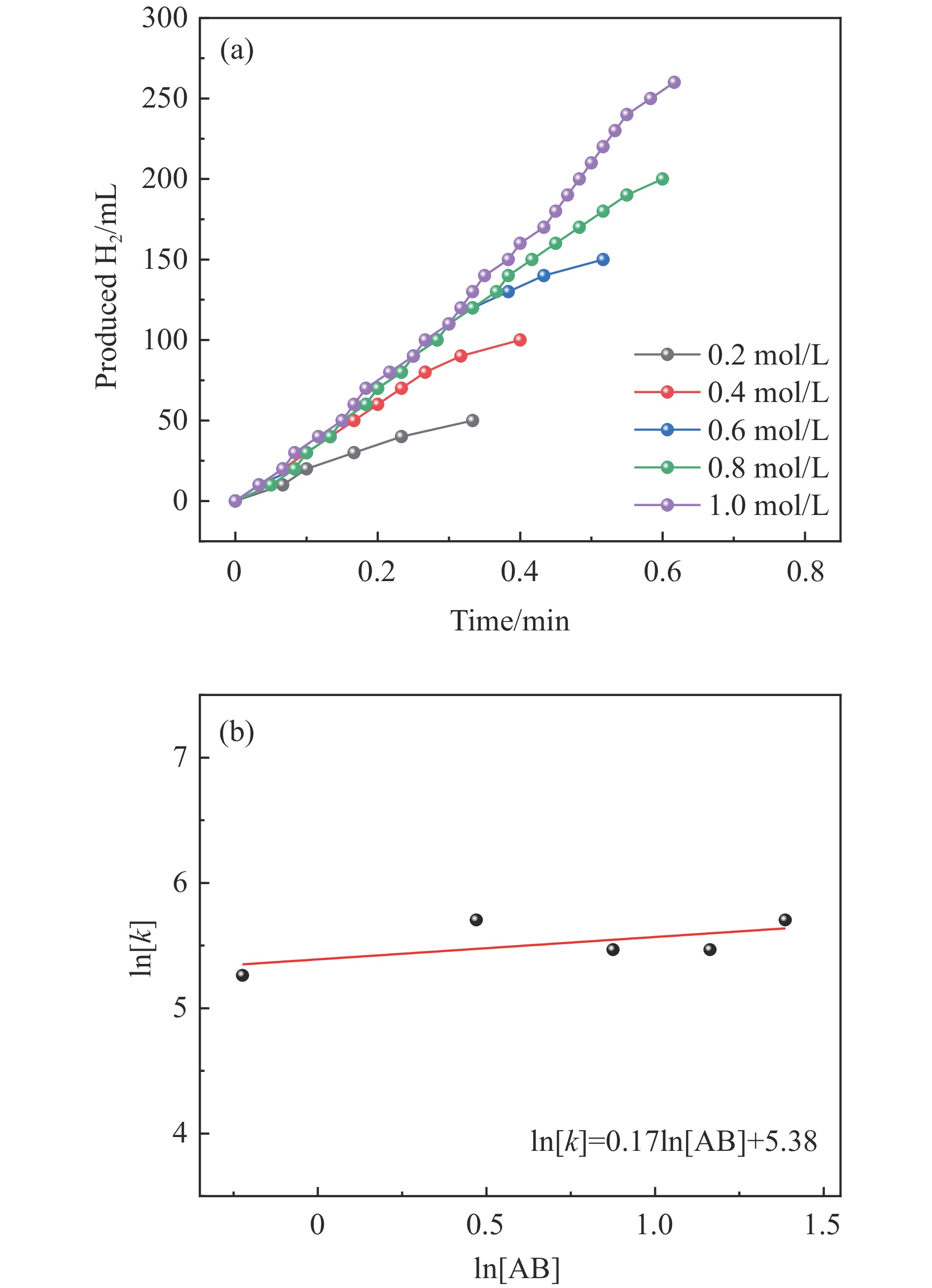
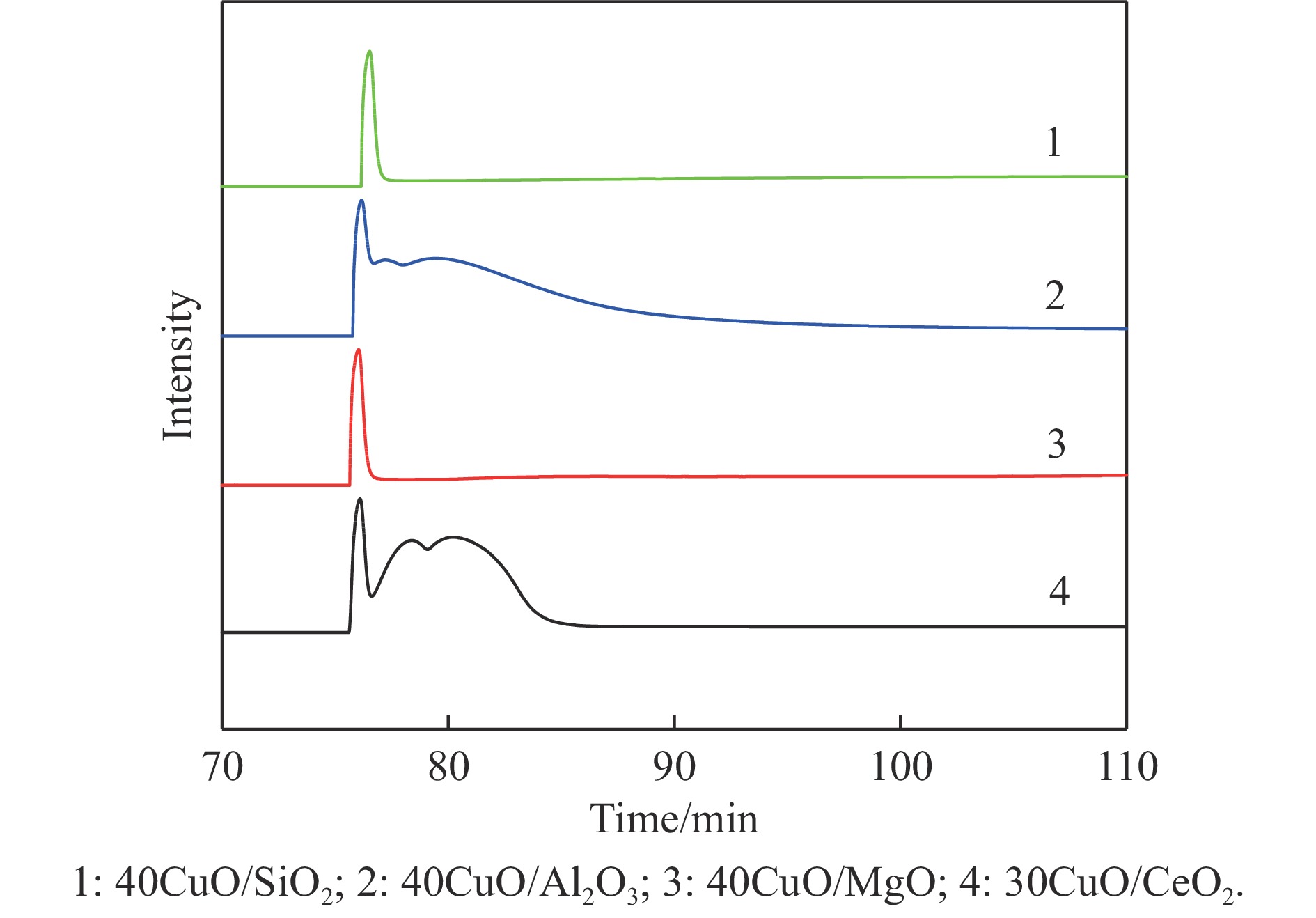
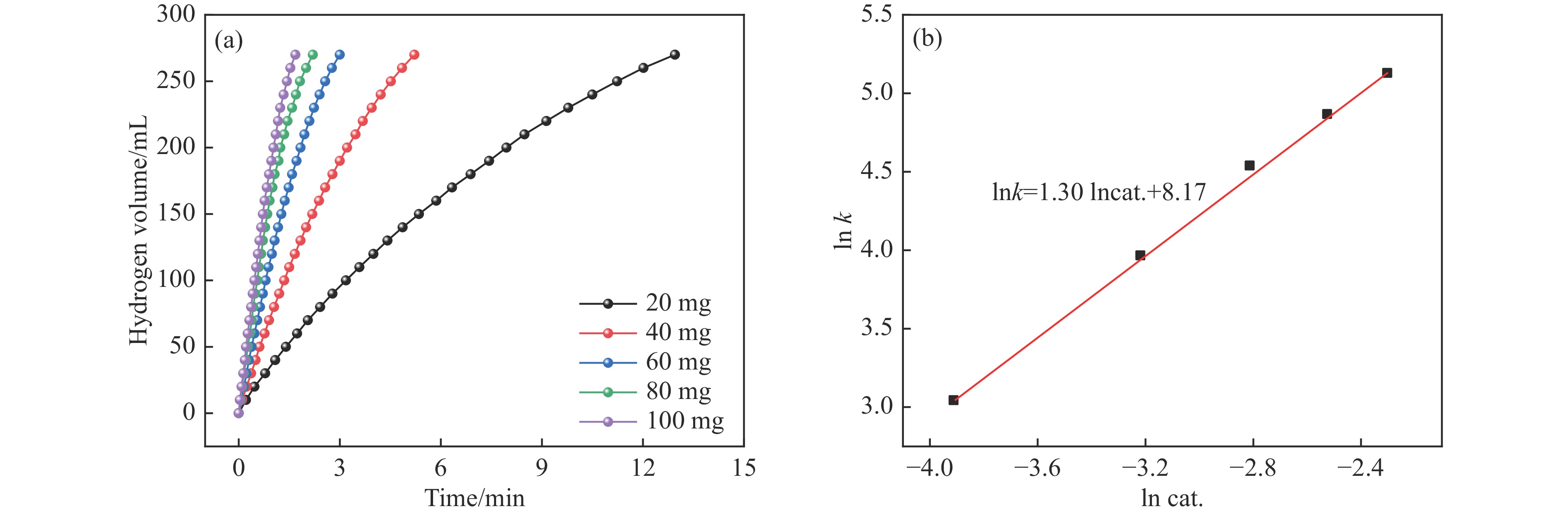
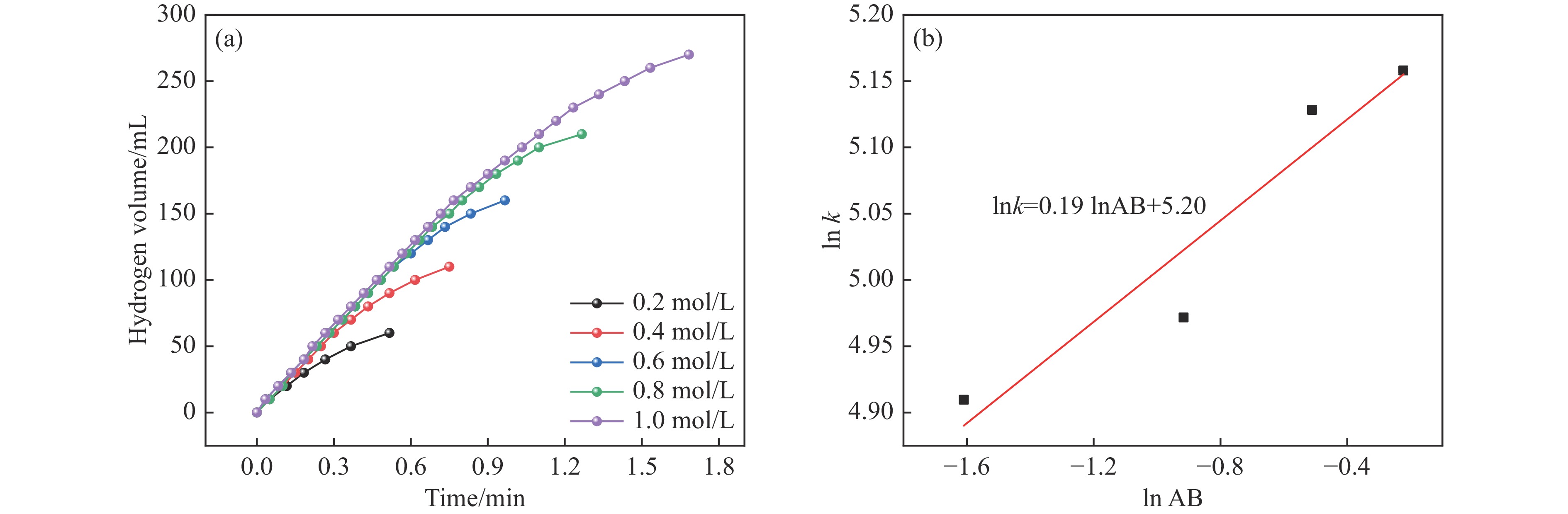
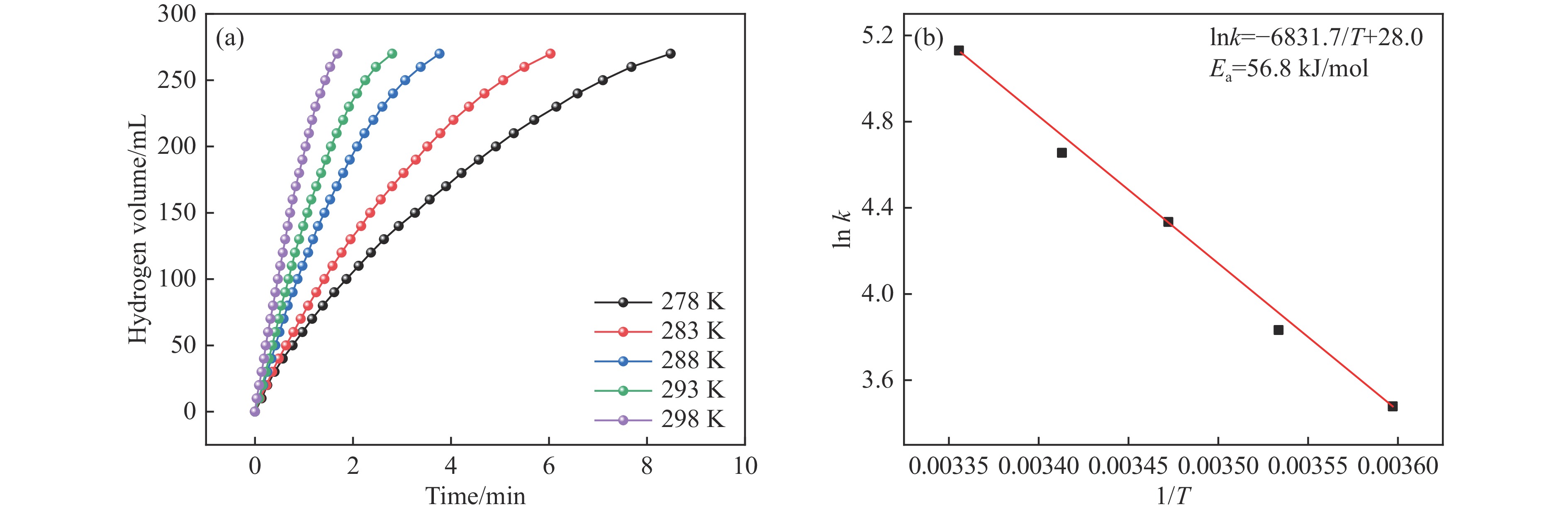
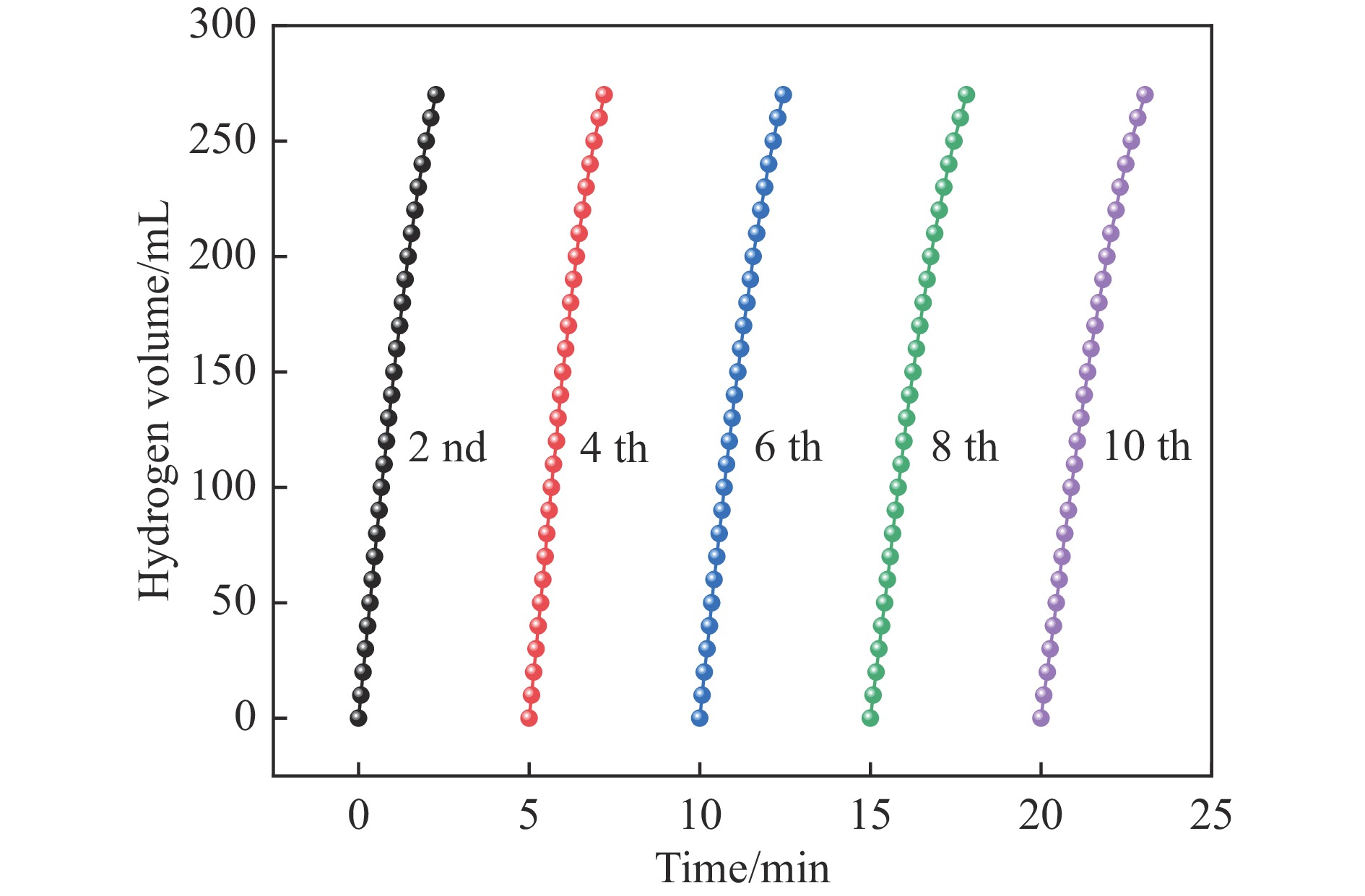
氨硼烷(NH3BH3,AB)是一种理想的制氢原料,具有较高的储氢能力。本研究在氮气气氛下高温碳化Ni/Cu-ZIF前驱体制备了一种含氮炭材料(Ni0.6Cu0.4O/NC)催化剂,并采用多种表征方法对所制备催化剂的微观结构以及组成成分进行了研究。此外,通过控制变量法探究了催化剂的催化性能以及变化规律。研究结果表明,Ni0.6Cu0.4O/NC催化AB水解制氢的活化能(Ea)为56.8 kJ/mol,TOF值高达1572.2 h−1。该催化剂催化AB水解制氢速率对于AB自身浓度可近似看作零级反应,而相对于催化剂的用量可近似看作一级反应。催化剂经过10次循环后仍然保持良好的催化活性,表明其具有良好的稳定性。
氨硼烷(NH3BH3,AB)是一种理想的制氢原料,具有较高的储氢能力。本研究在氮气气氛下高温碳化Ni/Cu-ZIF前驱体制备了一种含氮炭材料(Ni0.6Cu0.4O/NC)催化剂,并采用多种表征方法对所制备催化剂的微观结构以及组成成分进行了研究。此外,通过控制变量法探究了催化剂的催化性能以及变化规律。研究结果表明,Ni0.6Cu0.4O/NC催化AB水解制氢的活化能(Ea)为56.8 kJ/mol,TOF值高达1572.2 h−1。该催化剂催化AB水解制氢速率对于AB自身浓度可近似看作零级反应,而相对于催化剂的用量可近似看作一级反应。催化剂经过10次循环后仍然保持良好的催化活性,表明其具有良好的稳定性。

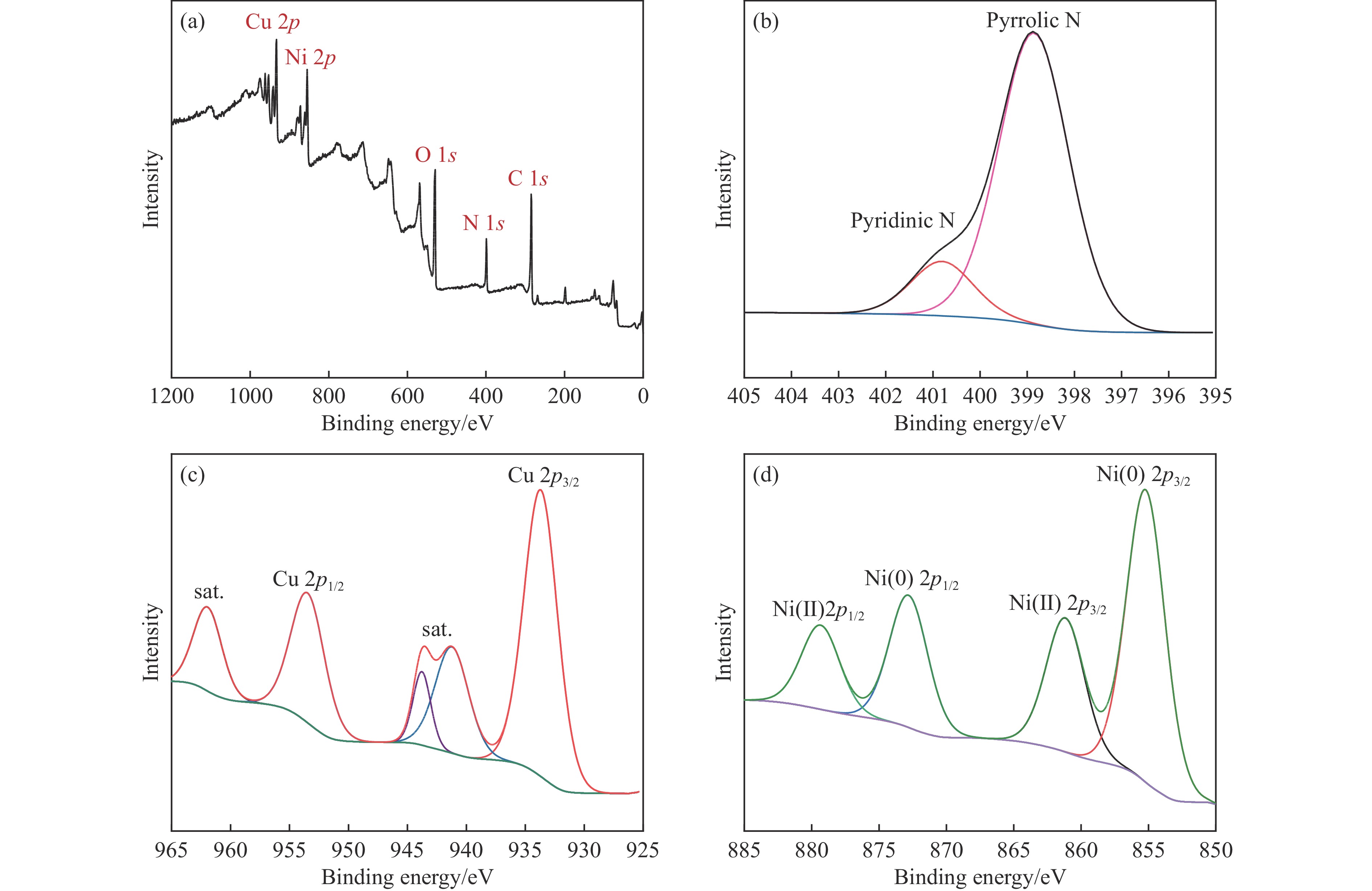
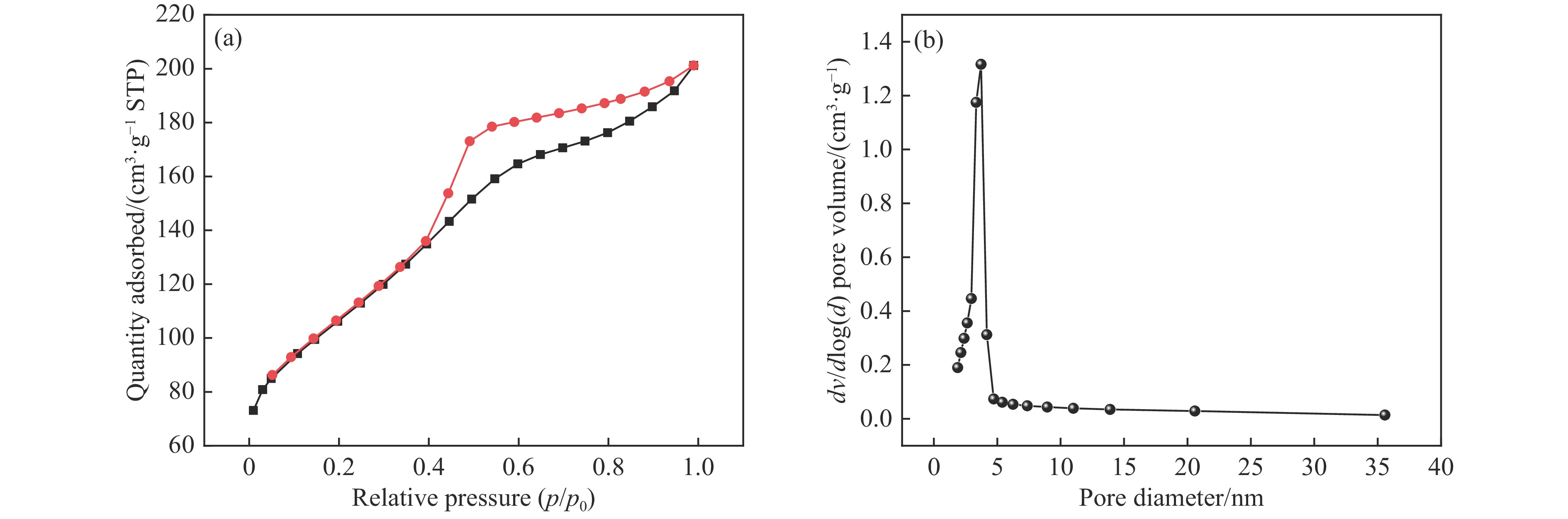
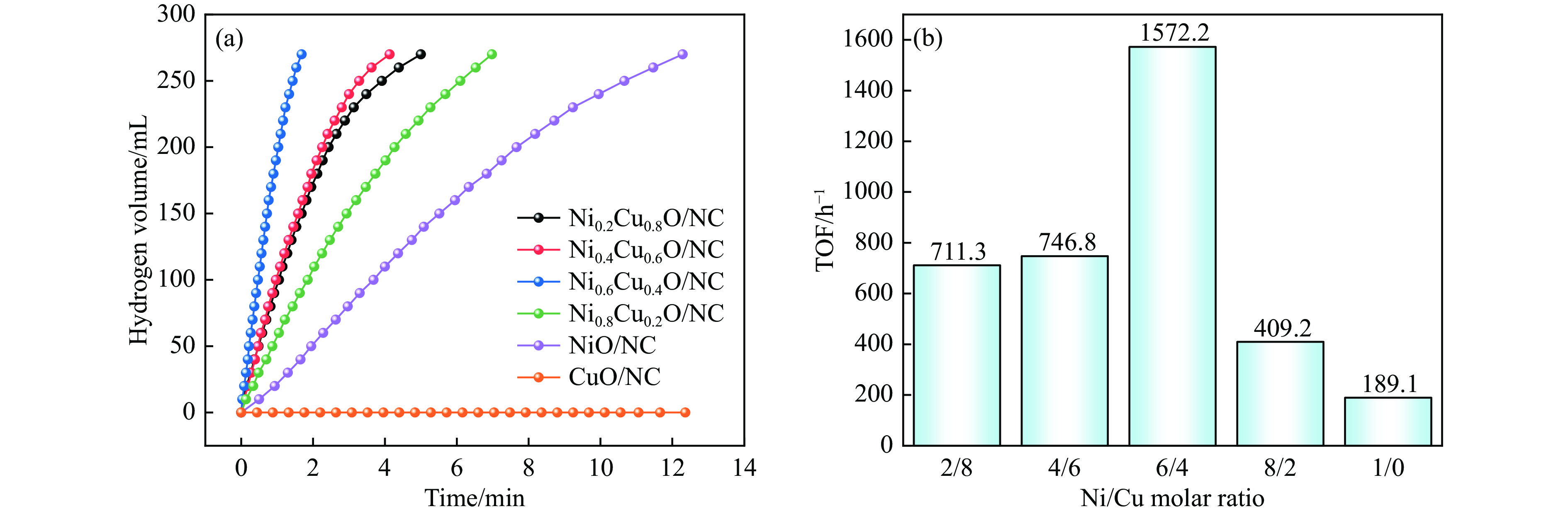

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2023088
摘要:
褐煤碳含量高且富含氧、氮等杂原子,是制备炭材料的重要原料。但由于褐煤可溶有机碳含量低,杂原子分配无规律,导致以褐煤为原料制备炭材料面临诸多挑战。因此,亟需实现褐煤的可溶化转化。本研究以氨水为溶剂,旨在温和条件下,同步实现昭通褐煤的可溶化和褐煤热溶物中氧和氮的调控。实验结果表明,在氨水浓度15%、温度160 ℃条件下反应3 h,热溶物收率最高为76.66%,昭通褐煤表现出良好的热溶效果。基于对热溶物的表征和分析,发现氨解在一定程度上改变了煤中的大分子结构,表现为氨基与羟基置换,或与部分羧基、羰基直接反应生成有机态氮。对比发现,原煤中氮元素赋存形态以季氮和吡咯氮为主,而可溶物中氮元素赋存形态以氨基氮和吡啶氮为主,表明褐煤氨解热溶过程产生了氨基或酰胺基。
褐煤碳含量高且富含氧、氮等杂原子,是制备炭材料的重要原料。但由于褐煤可溶有机碳含量低,杂原子分配无规律,导致以褐煤为原料制备炭材料面临诸多挑战。因此,亟需实现褐煤的可溶化转化。本研究以氨水为溶剂,旨在温和条件下,同步实现昭通褐煤的可溶化和褐煤热溶物中氧和氮的调控。实验结果表明,在氨水浓度15%、温度160 ℃条件下反应3 h,热溶物收率最高为76.66%,昭通褐煤表现出良好的热溶效果。基于对热溶物的表征和分析,发现氨解在一定程度上改变了煤中的大分子结构,表现为氨基与羟基置换,或与部分羧基、羰基直接反应生成有机态氮。对比发现,原煤中氮元素赋存形态以季氮和吡咯氮为主,而可溶物中氮元素赋存形态以氨基氮和吡啶氮为主,表明褐煤氨解热溶过程产生了氨基或酰胺基。











当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60406-2
摘要:
A series of spinel catalysts, including ZnFe2O4, MgFe2O4, CuFe2O4, and MnFe2O4, were prepared and applied to the Fischer-Tropsch synthesis (FTS). Zn, Mg, Cu and Mn easily form spinels with Fe. Among them, Zn and Mg can significantly maintain the spinel structure during the pretreatment and reaction, resulting in a low CO conversion. Cu and Mn are beneficial to the formation of iron carbide during the reaction, resulting in an apparent influence on FTS performance. ZnFe2O4 has little effect on the hydrocarbon distribution and the olefin/paraffin (O/P) ratio of C2−C4. MgFe2O4 exhibits low selectivity for C5+ hydrocarbons, and the selectivity of$ {\mathrm{C}}_2^=-{\mathrm{C}}_4^=\;$ and the O/P ratio of C2−C4 in the product are increased due to the alkaline effect of Mg. Cu can promote the carbonization of the catalyst, so that CuFe2O4 has higher activity. Meanwhile, CuFe2O4 can significantly improve the selectivity of C5+ hydrocarbons. Moreover, Cu can promote the dissociation and activation of H2, which is beneficial to the secondary hydrogenation of olefins, thereby reducing the selectivity of $ {\mathrm{C}}_2^=-{\mathrm{C}}_4^=\;$ and the O/P ratio of C2−C4. Mn promotes carbonization during the reaction, but MnFe2O4 has little effect on the chain growth of hydrocarbon. However, Mn can promote the generation of a certain amount of ε-Fe2C, which may explain the higher selectivity of $ {\mathrm{C}}_2^=-{\mathrm{C}}_4^=\;$ and the O/P ratio of C2−C4 for MnFe2O4. All spinel catalysts exhibit low CO2 selectivity, which meets the current green environmental protection requirements.
A series of spinel catalysts, including ZnFe2O4, MgFe2O4, CuFe2O4, and MnFe2O4, were prepared and applied to the Fischer-Tropsch synthesis (FTS). Zn, Mg, Cu and Mn easily form spinels with Fe. Among them, Zn and Mg can significantly maintain the spinel structure during the pretreatment and reaction, resulting in a low CO conversion. Cu and Mn are beneficial to the formation of iron carbide during the reaction, resulting in an apparent influence on FTS performance. ZnFe2O4 has little effect on the hydrocarbon distribution and the olefin/paraffin (O/P) ratio of C2−C4. MgFe2O4 exhibits low selectivity for C5+ hydrocarbons, and the selectivity of









当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60407-4
摘要:
基于密度泛函理论和经典过渡态理论,探究了石墨炭负载单原子Fe催化剂(Fe/G)异相还原NO的微观机理,并对催化剂失活原因进行分析。结果表明,基于E-R机理,NO还原反应依次经历了N2O形成与释放、N2形成与释放四个阶段。而基于L-H机理,NO还原反应主要经历了N2形成与释放两个阶段。在E-R机理作用下,NO分子以N,O-down结构吸附在Fe原子上发生的NO还原反应的控速步骤能垒值仅为15.5 kJ/mol,小于其余路径控速步骤能垒值。由能垒角度分析,Fe原子上残留的活性氧被还原的能垒值高于NO还原生成N2的能垒值。NO分解后残留在Fe原子表面的活性氧抑制了NO的吸附与还原,Fe原子活性位的缺失导致催化剂的失活,单原子Fe的存在促进了NO还原反应的进行。由动力学角度分析,随着反应温度的升高,NO还原速率较活性氧转移速率提升更为显著。
基于密度泛函理论和经典过渡态理论,探究了石墨炭负载单原子Fe催化剂(Fe/G)异相还原NO的微观机理,并对催化剂失活原因进行分析。结果表明,基于E-R机理,NO还原反应依次经历了N2O形成与释放、N2形成与释放四个阶段。而基于L-H机理,NO还原反应主要经历了N2形成与释放两个阶段。在E-R机理作用下,NO分子以N,O-down结构吸附在Fe原子上发生的NO还原反应的控速步骤能垒值仅为15.5 kJ/mol,小于其余路径控速步骤能垒值。由能垒角度分析,Fe原子上残留的活性氧被还原的能垒值高于NO还原生成N2的能垒值。NO分解后残留在Fe原子表面的活性氧抑制了NO的吸附与还原,Fe原子活性位的缺失导致催化剂的失活,单原子Fe的存在促进了NO还原反应的进行。由动力学角度分析,随着反应温度的升高,NO还原速率较活性氧转移速率提升更为显著。










当前状态:
, 最新更新时间: ,
doi: 10.1016/S1872-5813(23)60405-0
摘要:
Phenolic derivatives, crucial components of bio-oil, require thorough understanding of their electrocatalytic hydrogenation (ECH) properties for efficient bio-oil utilization. This study investigated guaiacol, a representative phenolic derivative in bio-oil, focusing on its ECH mechanism, conversion, and product selectivity under varied conditions (temperature: 40−80 °C, perchloric acid concentration: 0.2−1.0 mol/L, current intensity: ((−10)−150 mA). Additionally, this study also explored the influence of intermediate products (2-methoxycyclohexanone and cyclohexanone) on both the conversion rate and the selectivity of the products. The experiment had revealed that guaiacol's ECH conversion rate improved with higher temperature and current intensity, whereas an increase in perchloric acid concentration negatively affected the conversion. Significantly, the presence of intermediate products, especially 2-methoxycyclohexanone, markedly enhanced the ECH conversion of guaiacol. Investigating further into the ECH mechanism of other phenolic derivatives, including phenol, pyrocatechol, guaiacol eugenol, and vanillin, as well as their combination, revealed a trend where conversion rates inversely correlated with the complexity of the functional groups on the benzene ring. Specifically, phenol, with its simpler structure, showed the highest conversion rate at 89.34%, in stark contrast to vanillin which, owing to its more complex structure, exhibited the lowest at 46.79%. In our multi-component mixture studies, it was observed that synergistic and competitive interactions significantly alter ECH conversion rates, with some mixtures showing enhanced conversion rate indicative of synergistic effects.
Phenolic derivatives, crucial components of bio-oil, require thorough understanding of their electrocatalytic hydrogenation (ECH) properties for efficient bio-oil utilization. This study investigated guaiacol, a representative phenolic derivative in bio-oil, focusing on its ECH mechanism, conversion, and product selectivity under varied conditions (temperature: 40−80 °C, perchloric acid concentration: 0.2−1.0 mol/L, current intensity: ((−10)−150 mA). Additionally, this study also explored the influence of intermediate products (2-methoxycyclohexanone and cyclohexanone) on both the conversion rate and the selectivity of the products. The experiment had revealed that guaiacol's ECH conversion rate improved with higher temperature and current intensity, whereas an increase in perchloric acid concentration negatively affected the conversion. Significantly, the presence of intermediate products, especially 2-methoxycyclohexanone, markedly enhanced the ECH conversion of guaiacol. Investigating further into the ECH mechanism of other phenolic derivatives, including phenol, pyrocatechol, guaiacol eugenol, and vanillin, as well as their combination, revealed a trend where conversion rates inversely correlated with the complexity of the functional groups on the benzene ring. Specifically, phenol, with its simpler structure, showed the highest conversion rate at 89.34%, in stark contrast to vanillin which, owing to its more complex structure, exhibited the lowest at 46.79%. In our multi-component mixture studies, it was observed that synergistic and competitive interactions significantly alter ECH conversion rates, with some mixtures showing enhanced conversion rate indicative of synergistic effects.






当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024003
摘要:
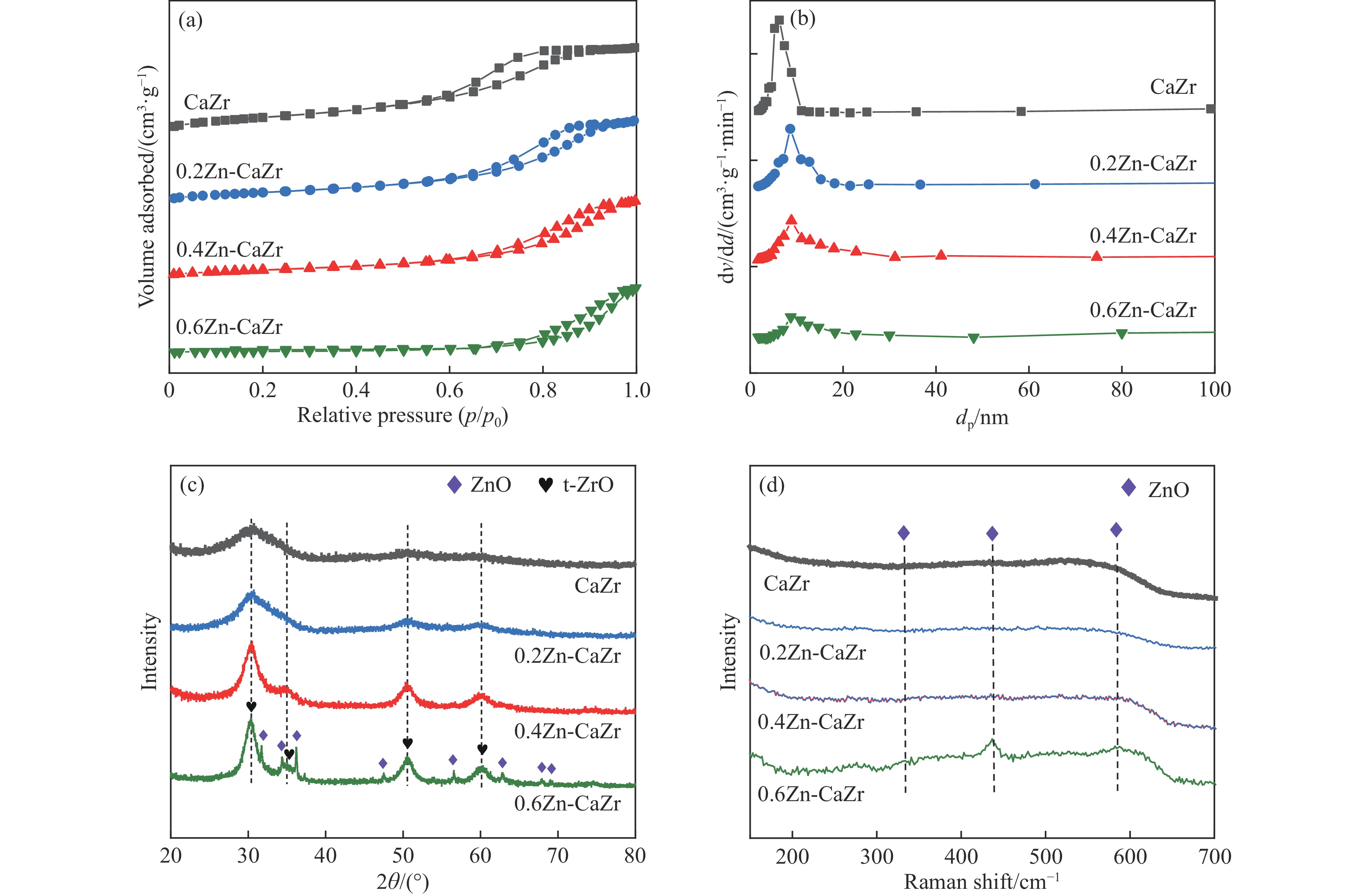
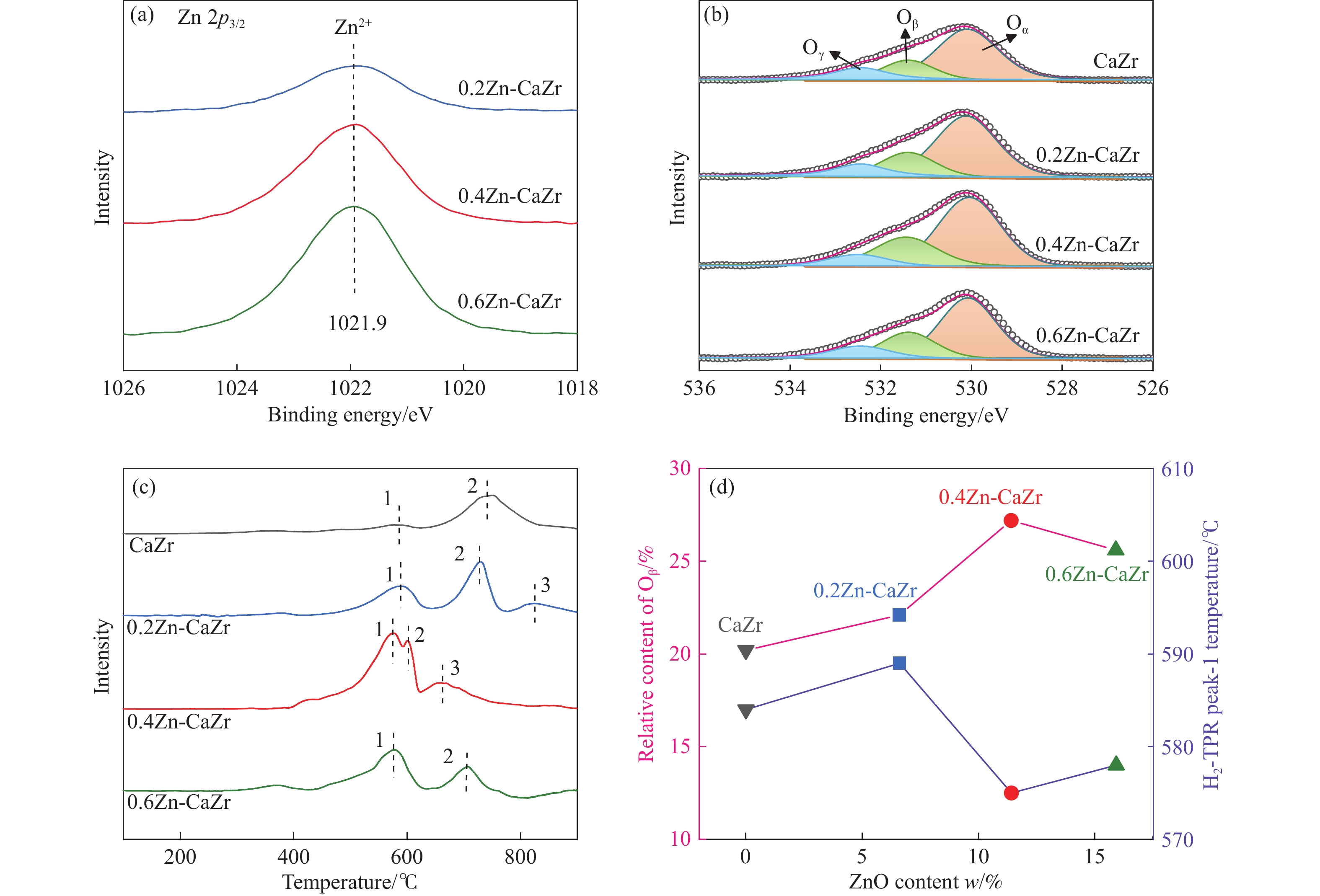
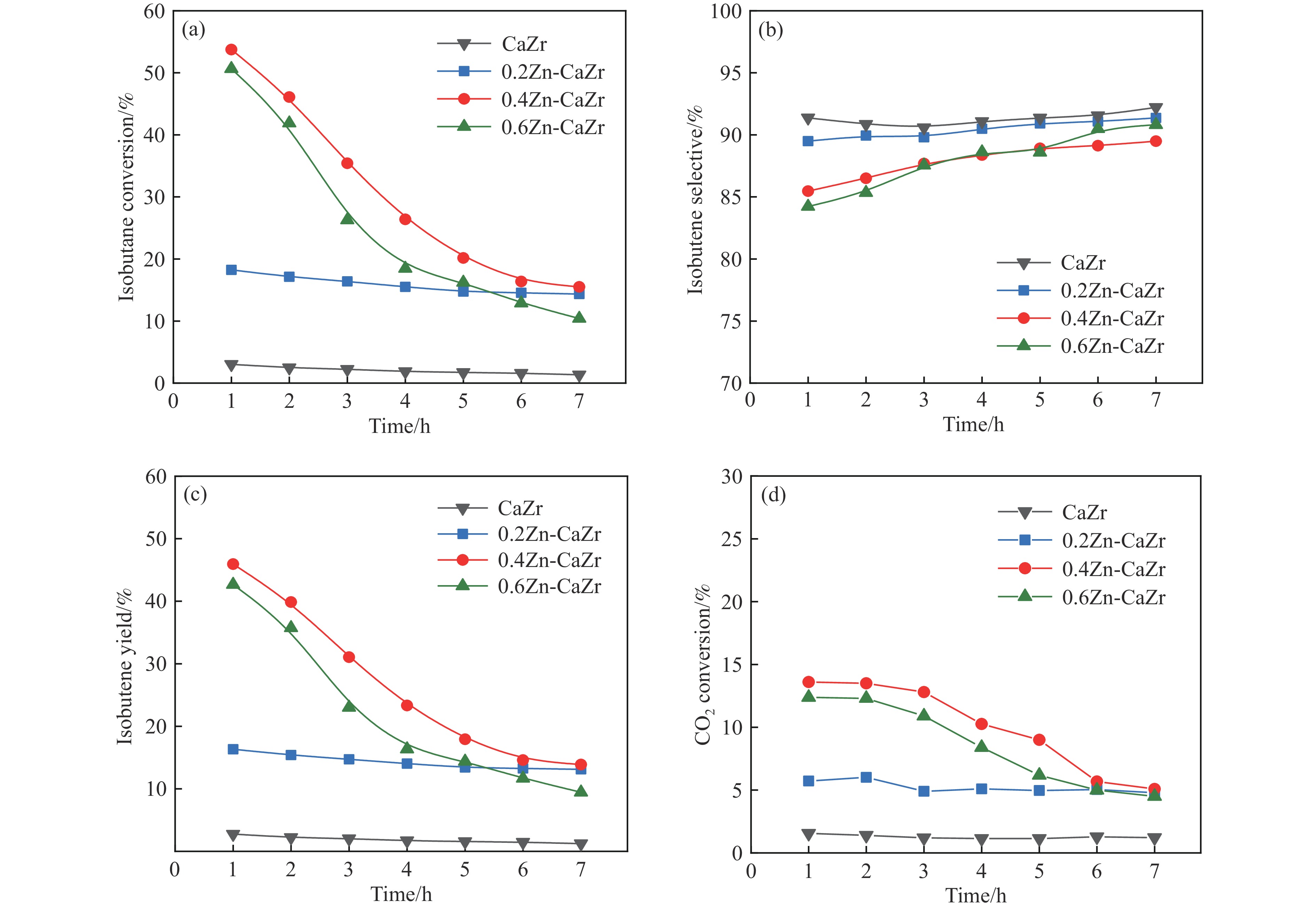
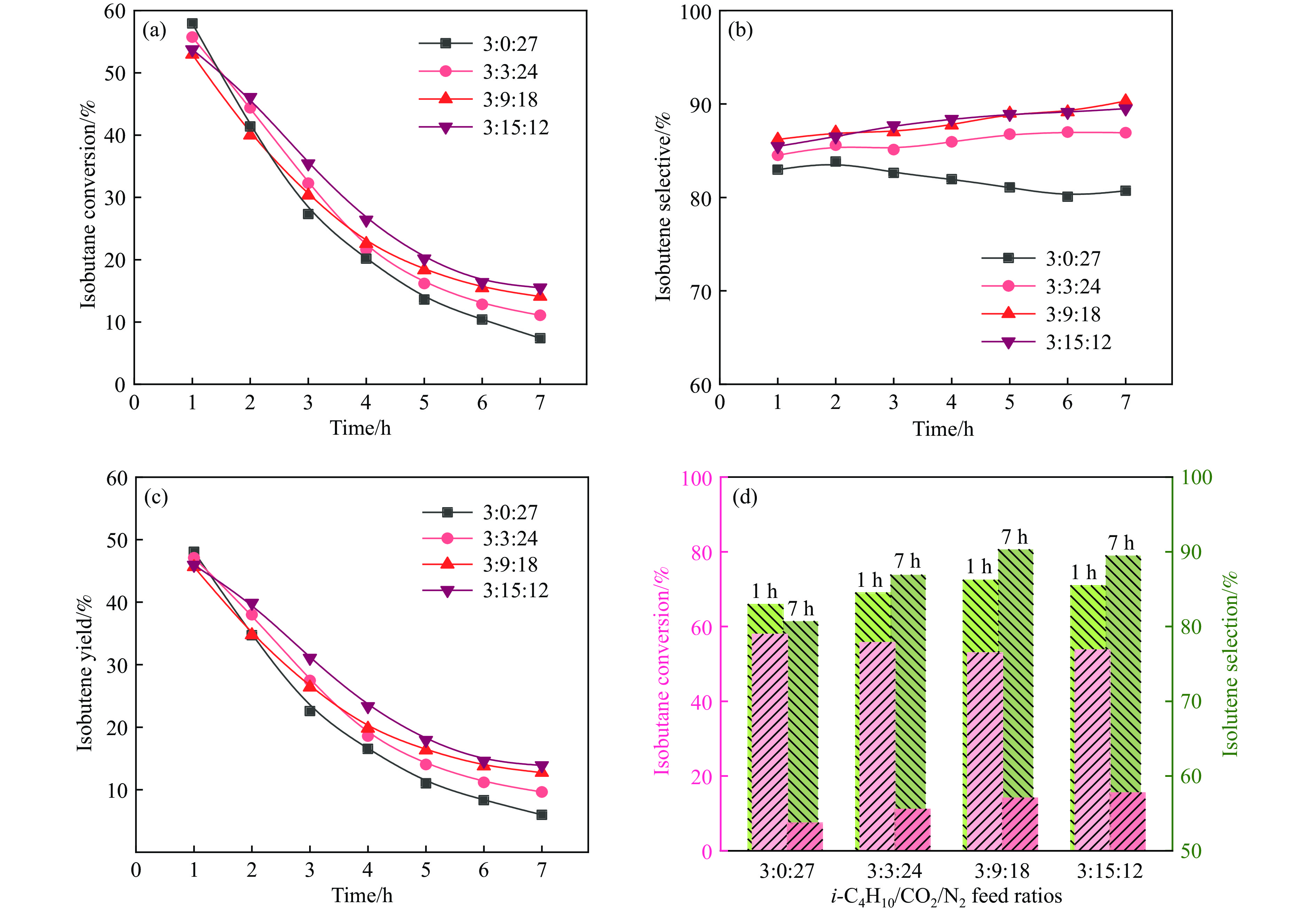
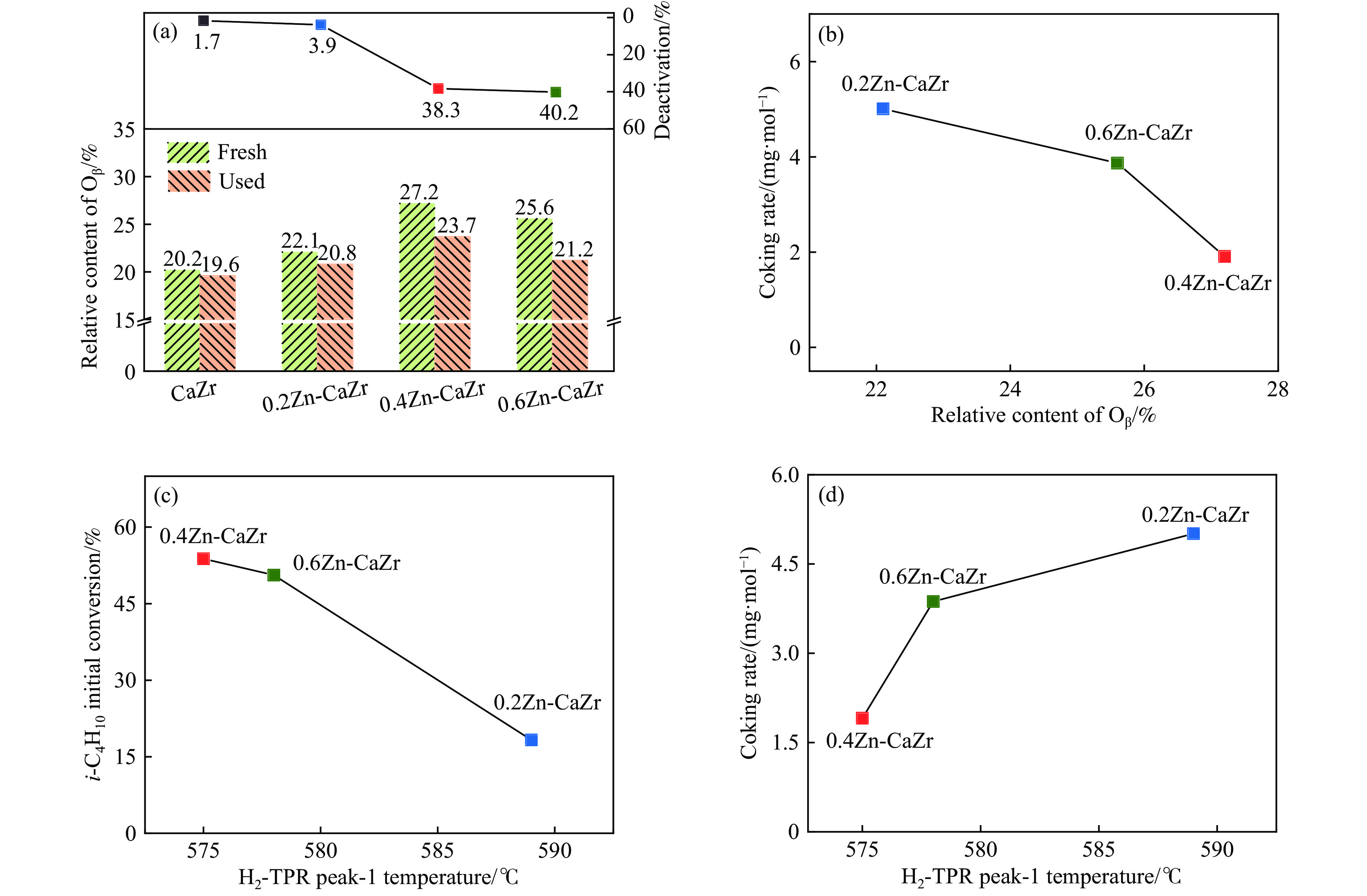
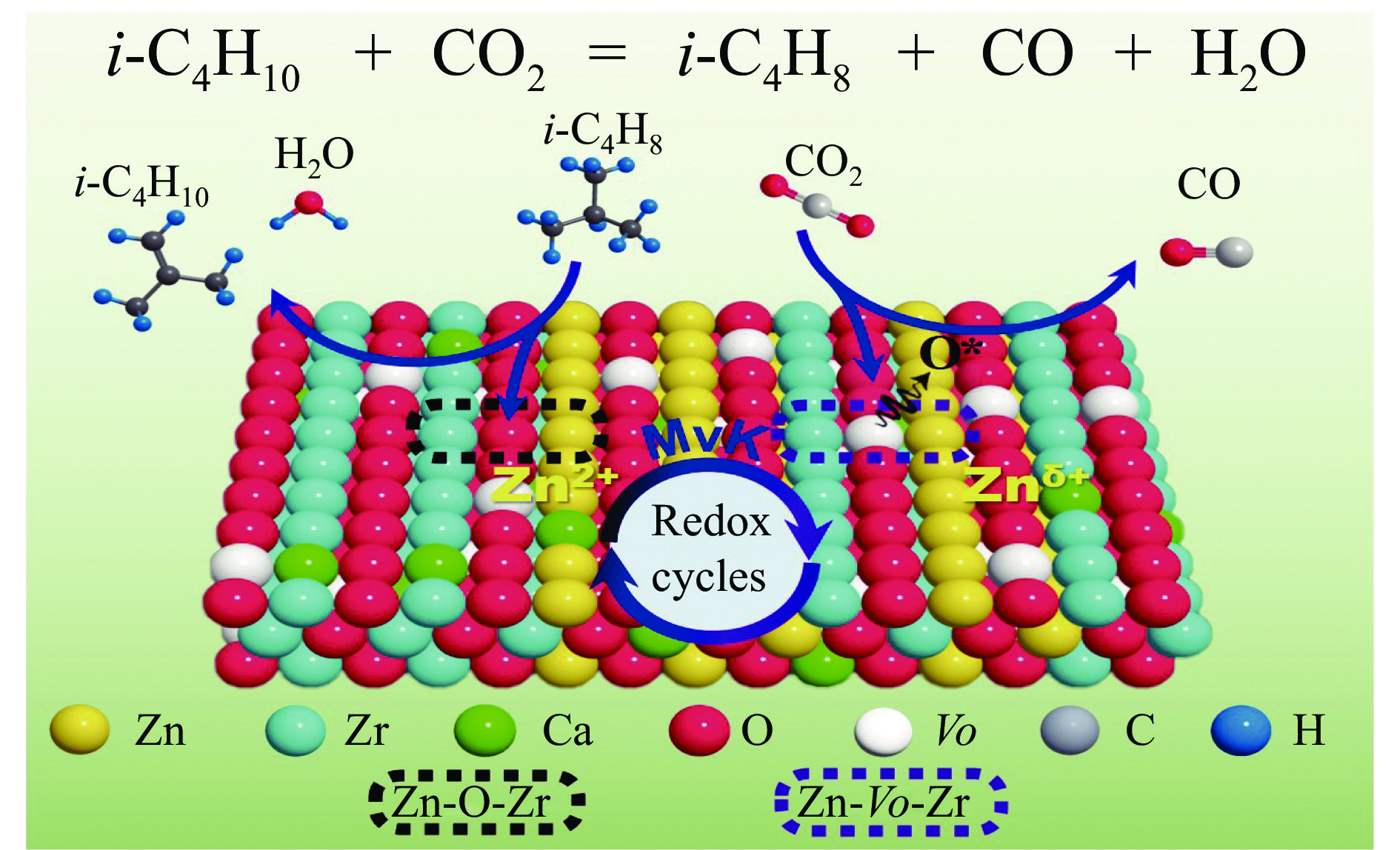
CO2介导的异丁烷氧化脱氢制异丁烯技术(CO2-BDH)是一种低碳环保的脱氢工艺,但目前尚缺乏绿色高效的催化剂,因此其工业化仍面临挑战。本文采用一锅式共沉淀法制备了xZn-CaZr固溶体催化剂并将其应用于CO2-BDH反应,通过多种手段探明该系列催化剂的理化性质并结合催化性能阐述其构效关系及表面氧化还原机制。研究表明,xZn-CaZr催化剂在Zn含量为6−12%的情况下形成了Zn物种高度分散的固溶体结构,且氧缺陷的数量与Zn的含量几乎成正比。在xZn-CaZr催化剂上,晶格氧的数量和氧迁移率是决定催化性能的关键因素,其中0.4Zn-CaZr催化剂展示出最佳的催化活性,而0.2Zn-CaZr催化剂展示出最佳的反应稳定性。该研究为进一步开发绿色高性能的CO2-BDH催化剂提供了参考价值。
CO2介导的异丁烷氧化脱氢制异丁烯技术(CO2-BDH)是一种低碳环保的脱氢工艺,但目前尚缺乏绿色高效的催化剂,因此其工业化仍面临挑战。本文采用一锅式共沉淀法制备了xZn-CaZr固溶体催化剂并将其应用于CO2-BDH反应,通过多种手段探明该系列催化剂的理化性质并结合催化性能阐述其构效关系及表面氧化还原机制。研究表明,xZn-CaZr催化剂在Zn含量为6−12%的情况下形成了Zn物种高度分散的固溶体结构,且氧缺陷的数量与Zn的含量几乎成正比。在xZn-CaZr催化剂上,晶格氧的数量和氧迁移率是决定催化性能的关键因素,其中0.4Zn-CaZr催化剂展示出最佳的催化活性,而0.2Zn-CaZr催化剂展示出最佳的反应稳定性。该研究为进一步开发绿色高性能的CO2-BDH催化剂提供了参考价值。

当前状态:
, 最新更新时间: ,
doi: 10.19906/j.cnki.JFCT.2024001
摘要:
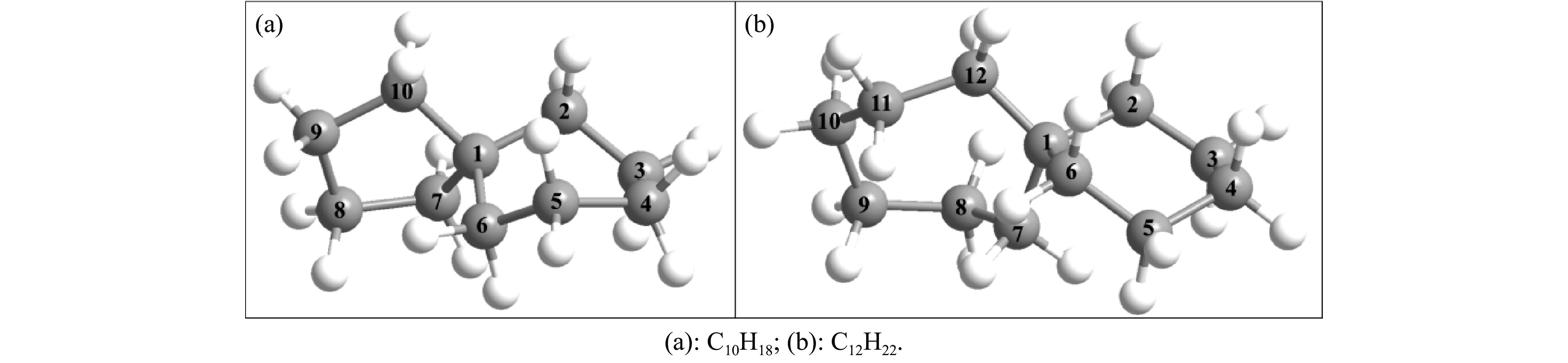
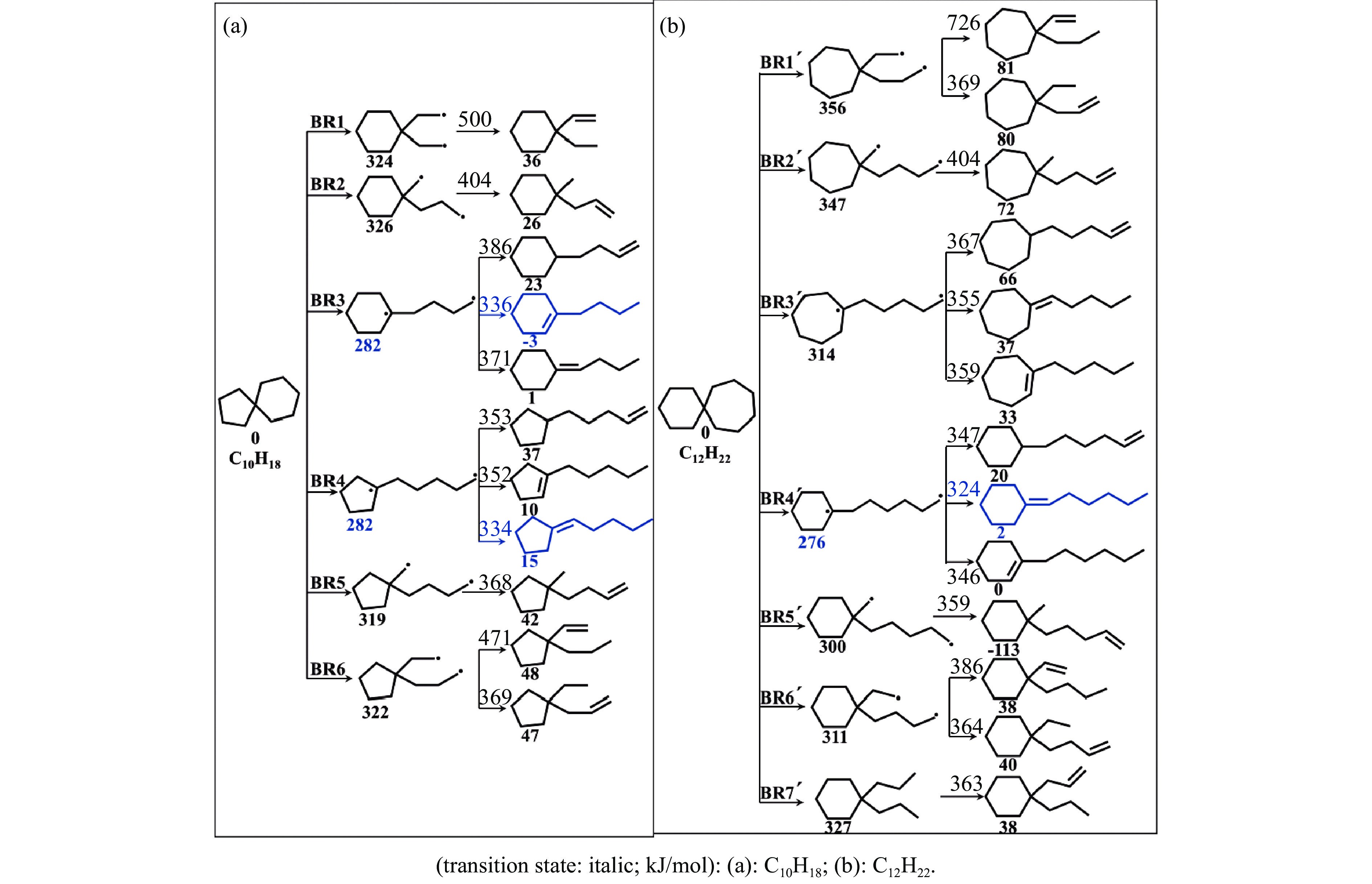
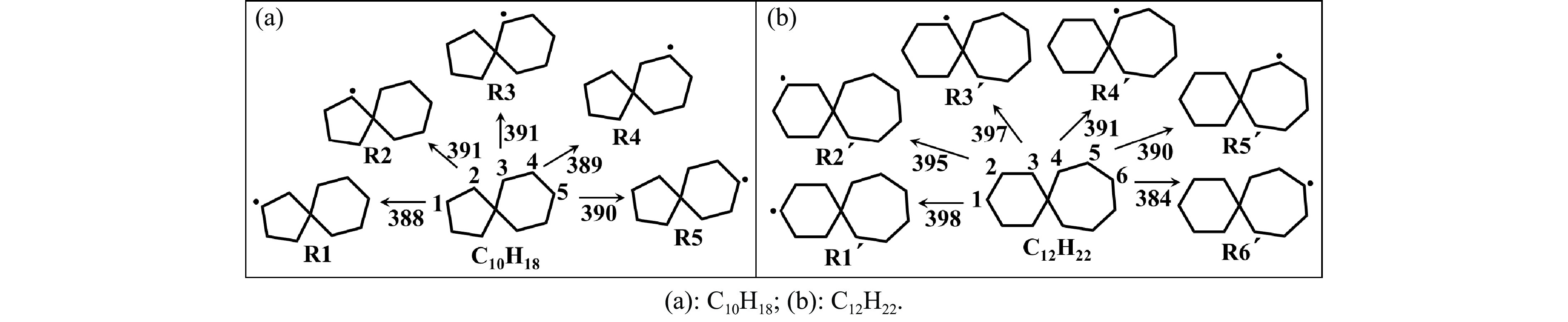
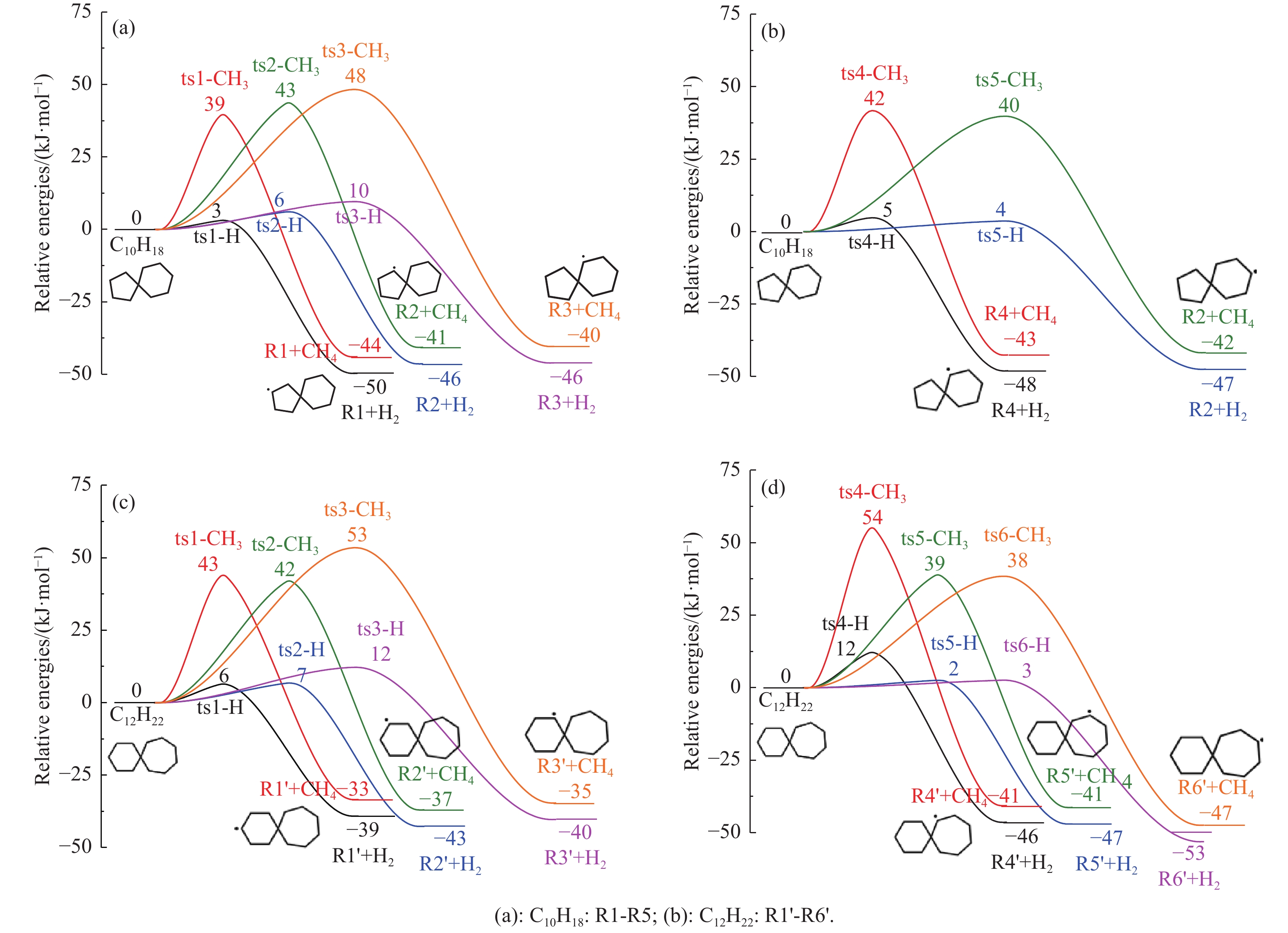
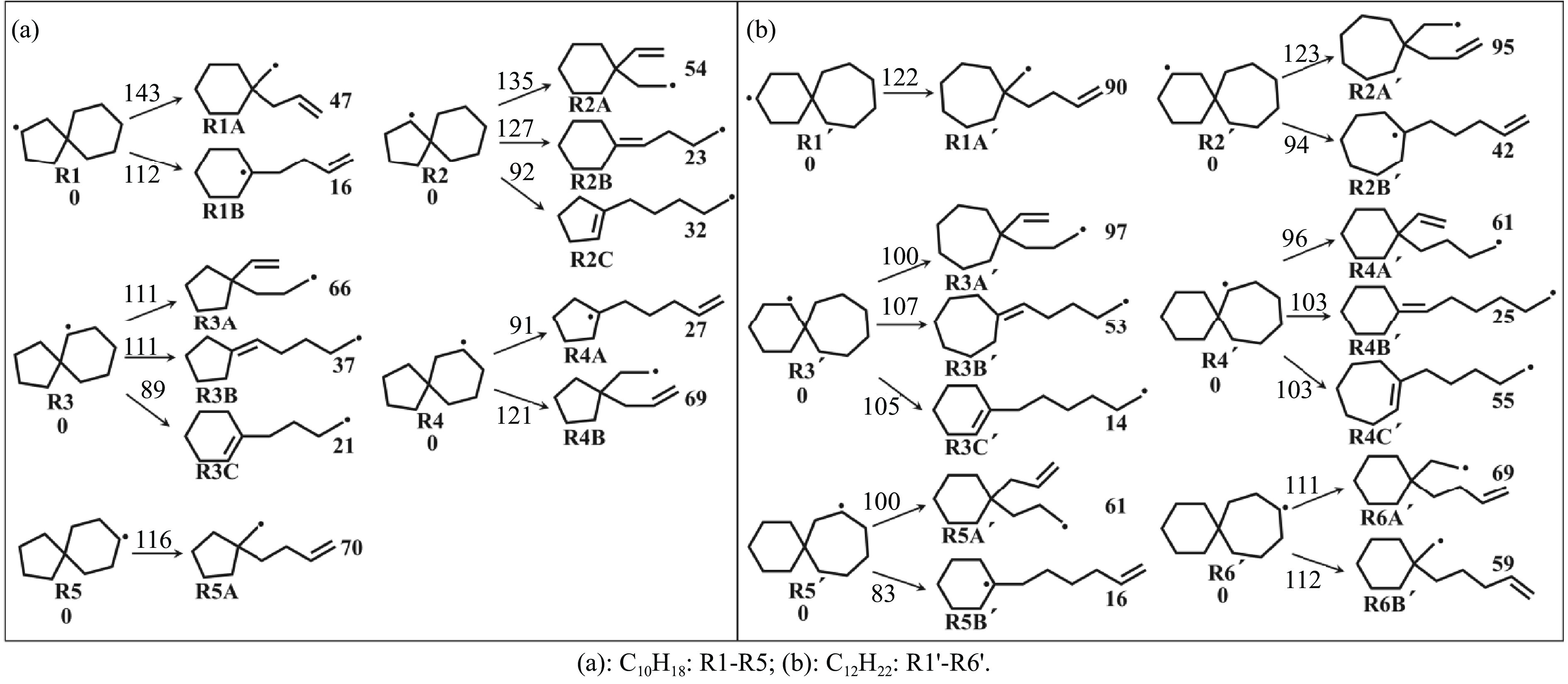
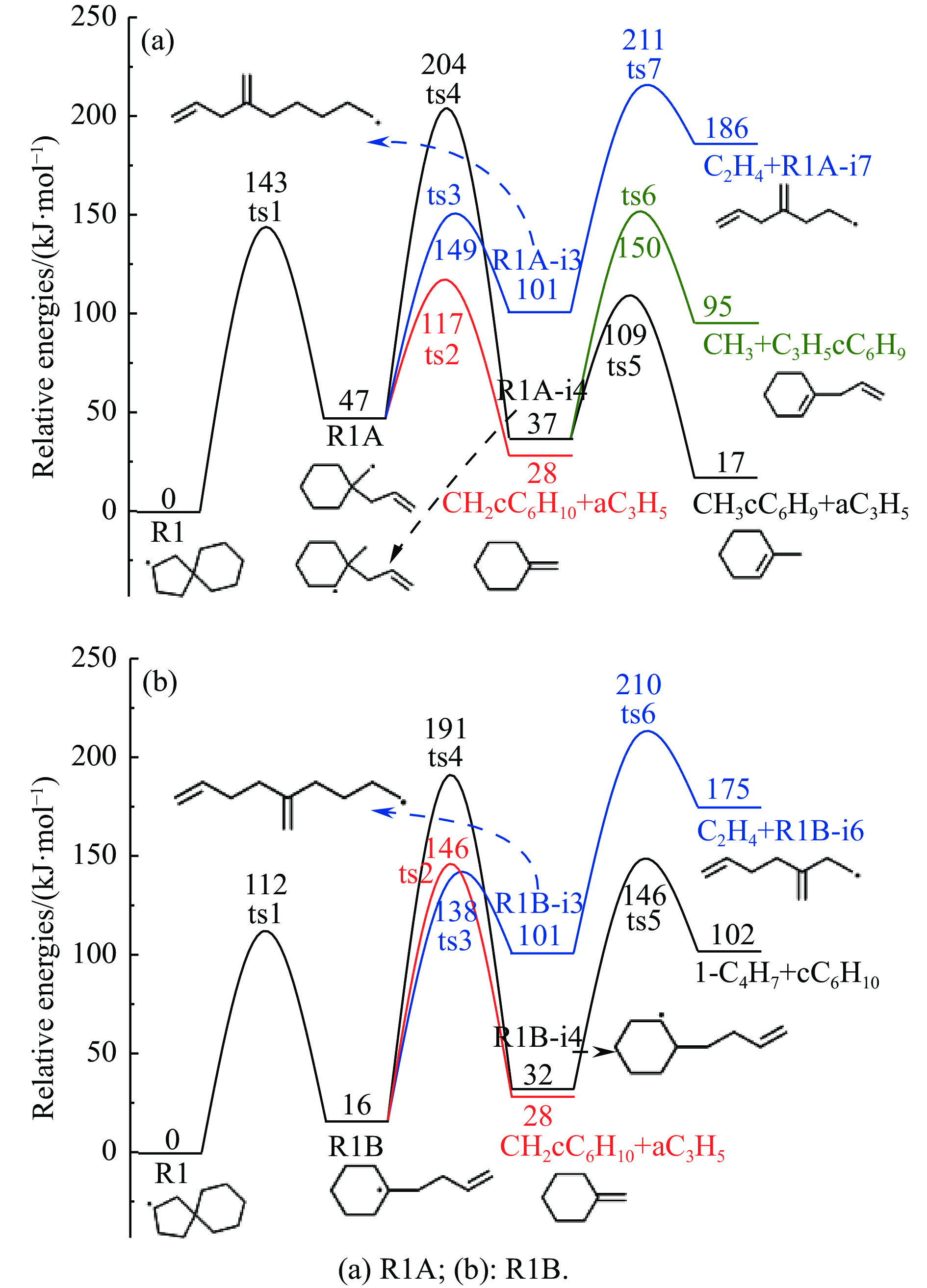
采用B3LYP/6-311++G(d,p)和反应性分子动力学方法,对螺[4,5]-癸烷(C10H18)和螺[5,6]-十二烷(C10H22)的热解机理进行研究,揭示不同碳环结构和尺寸效应对燃料初始分解反应活性及小分子和芳烃产物生成行为的影响。结果表明,两种螺环烷烃燃料初始分解路径相似,均通过单分子碳碳键解离发生开环异构反应和小分子自由基进攻燃料母体的氢提取反应而消耗。相较于C10H18,C12H22中分子张力更大的七元环使速控步碳碳键及碳氢键能更低,导致燃料呈现出更低的初始分解温度和更高的反应活性。两种螺环燃料初始分解产生的自由基进一步影响了C1−C7小分子烃类和环烯产物的生成。其中,乙烯的生成始终占据主导地位。由于螺环尺寸效应的影响,链烃和环烯烃的生成表现出明显的结构差异性。对于C10H18分解而言,可能生成大量的五元环烯产物,包括环戊二烯、环戊烯、富烯、甲基环戊二烯和甲基环戊烯;而C12H22中更大的七元环结构,将生成对应的七元环烯产物(环庚烯、亚甲基环庚烷)。
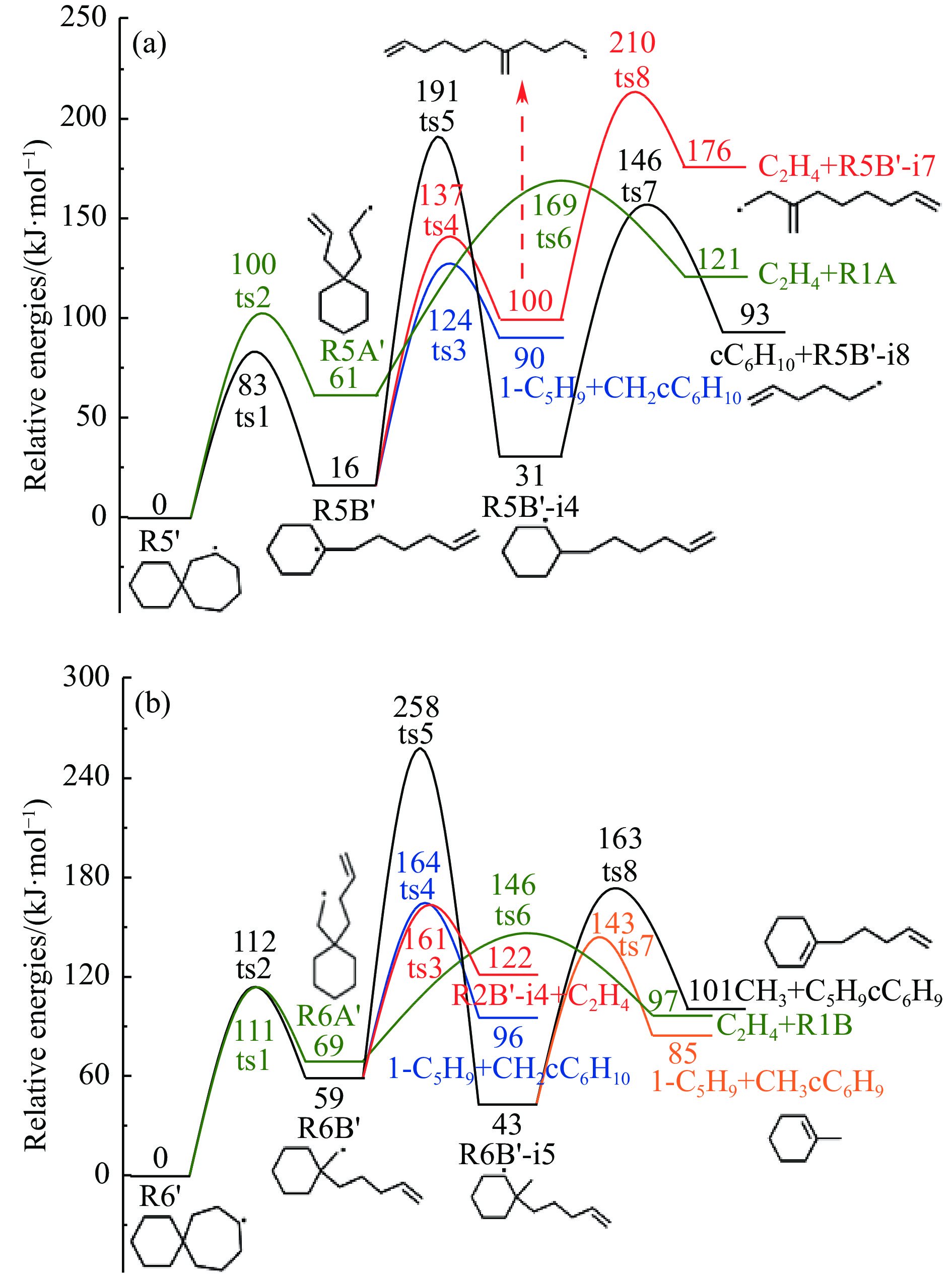
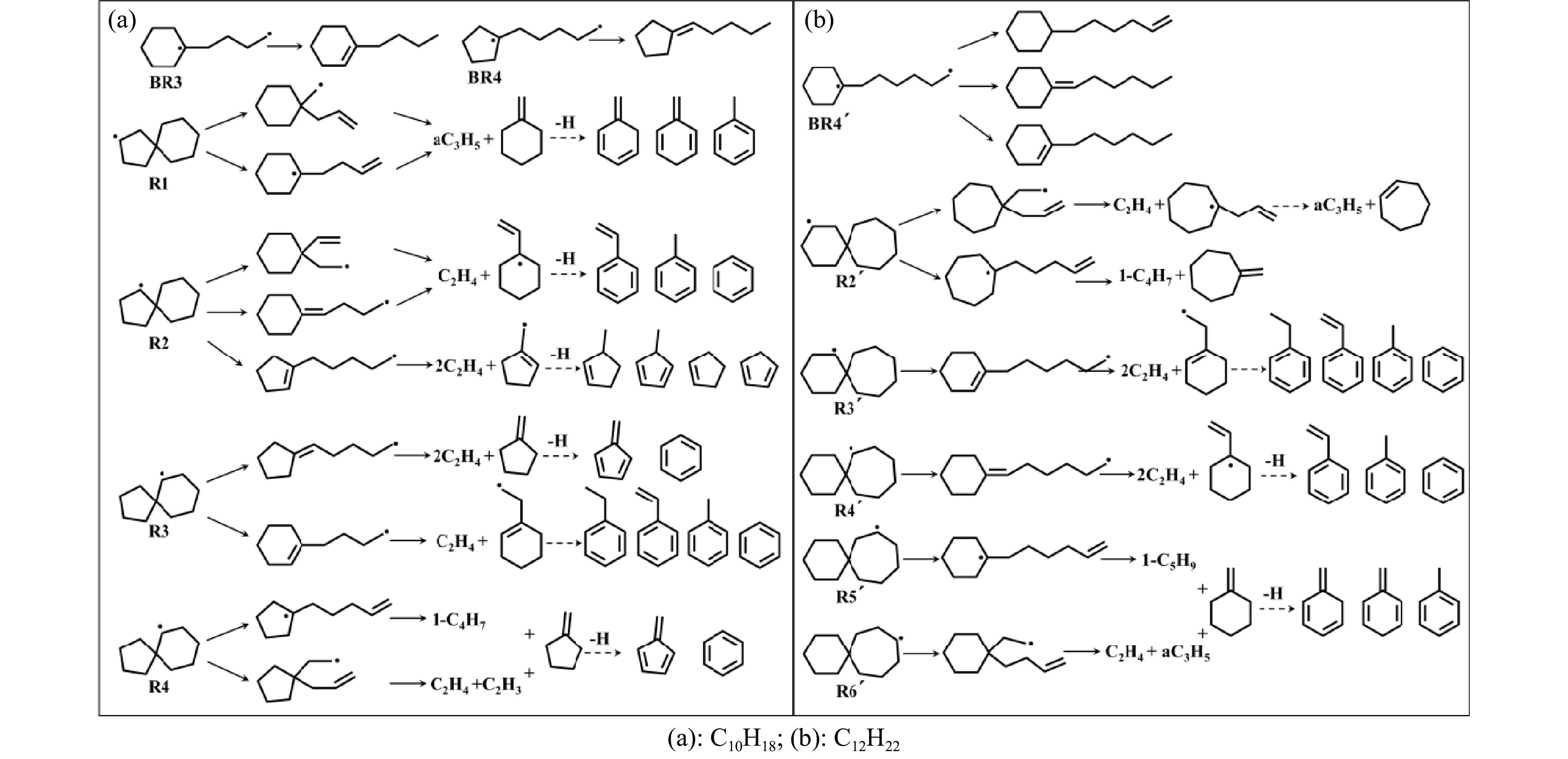
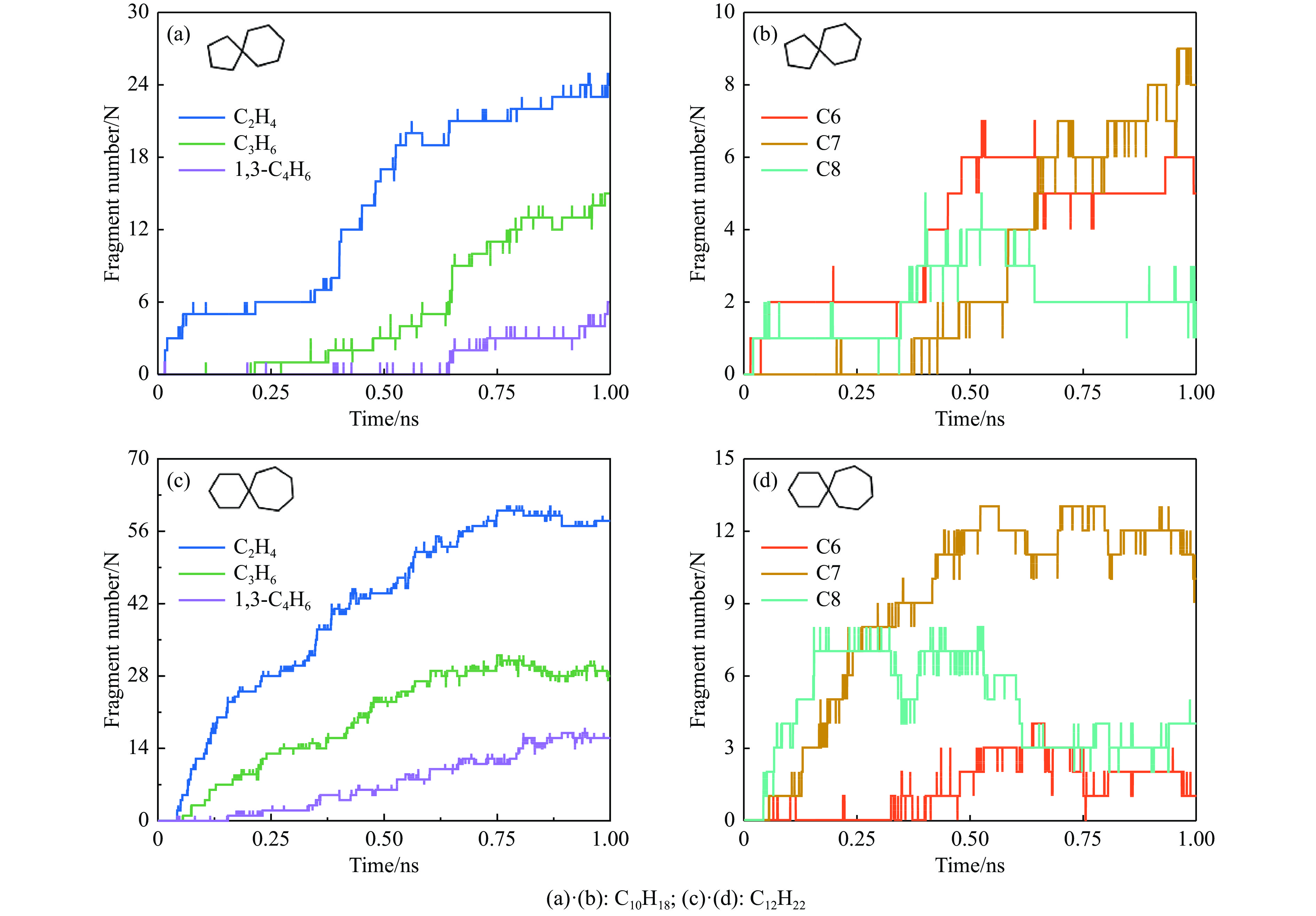
采用B3LYP/6-311++G(d,p)和反应性分子动力学方法,对螺[4,5]-癸烷(C10H18)和螺[5,6]-十二烷(C10H22)的热解机理进行研究,揭示不同碳环结构和尺寸效应对燃料初始分解反应活性及小分子和芳烃产物生成行为的影响。结果表明,两种螺环烷烃燃料初始分解路径相似,均通过单分子碳碳键解离发生开环异构反应和小分子自由基进攻燃料母体的氢提取反应而消耗。相较于C10H18,C12H22中分子张力更大的七元环使速控步碳碳键及碳氢键能更低,导致燃料呈现出更低的初始分解温度和更高的反应活性。两种螺环燃料初始分解产生的自由基进一步影响了C1−C7小分子烃类和环烯产物的生成。其中,乙烯的生成始终占据主导地位。由于螺环尺寸效应的影响,链烃和环烯烃的生成表现出明显的结构差异性。对于C10H18分解而言,可能生成大量的五元环烯产物,包括环戊二烯、环戊烯、富烯、甲基环戊二烯和甲基环戊烯;而C12H22中更大的七元环结构,将生成对应的七元环烯产物(环庚烯、亚甲基环庚烷)。

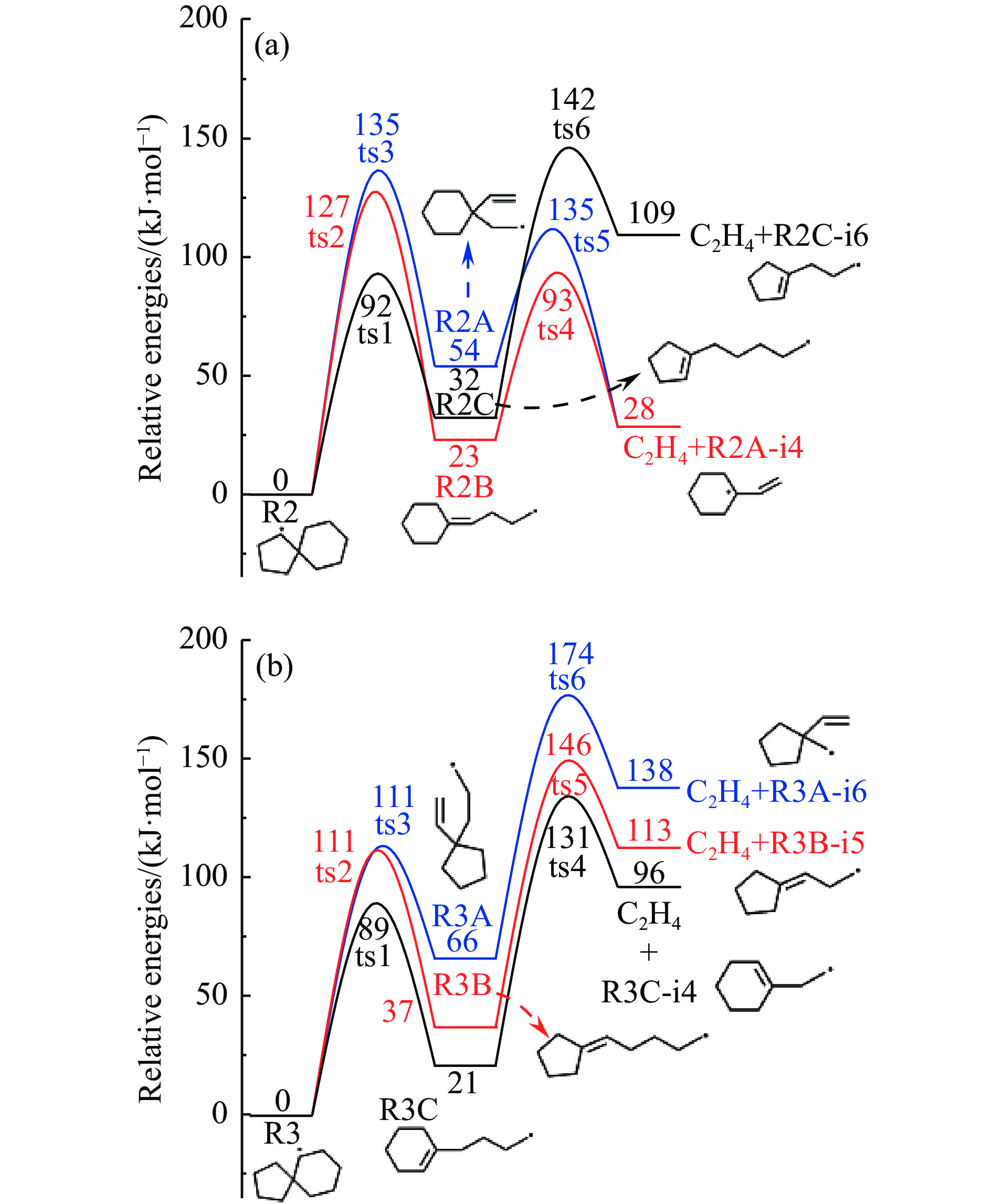
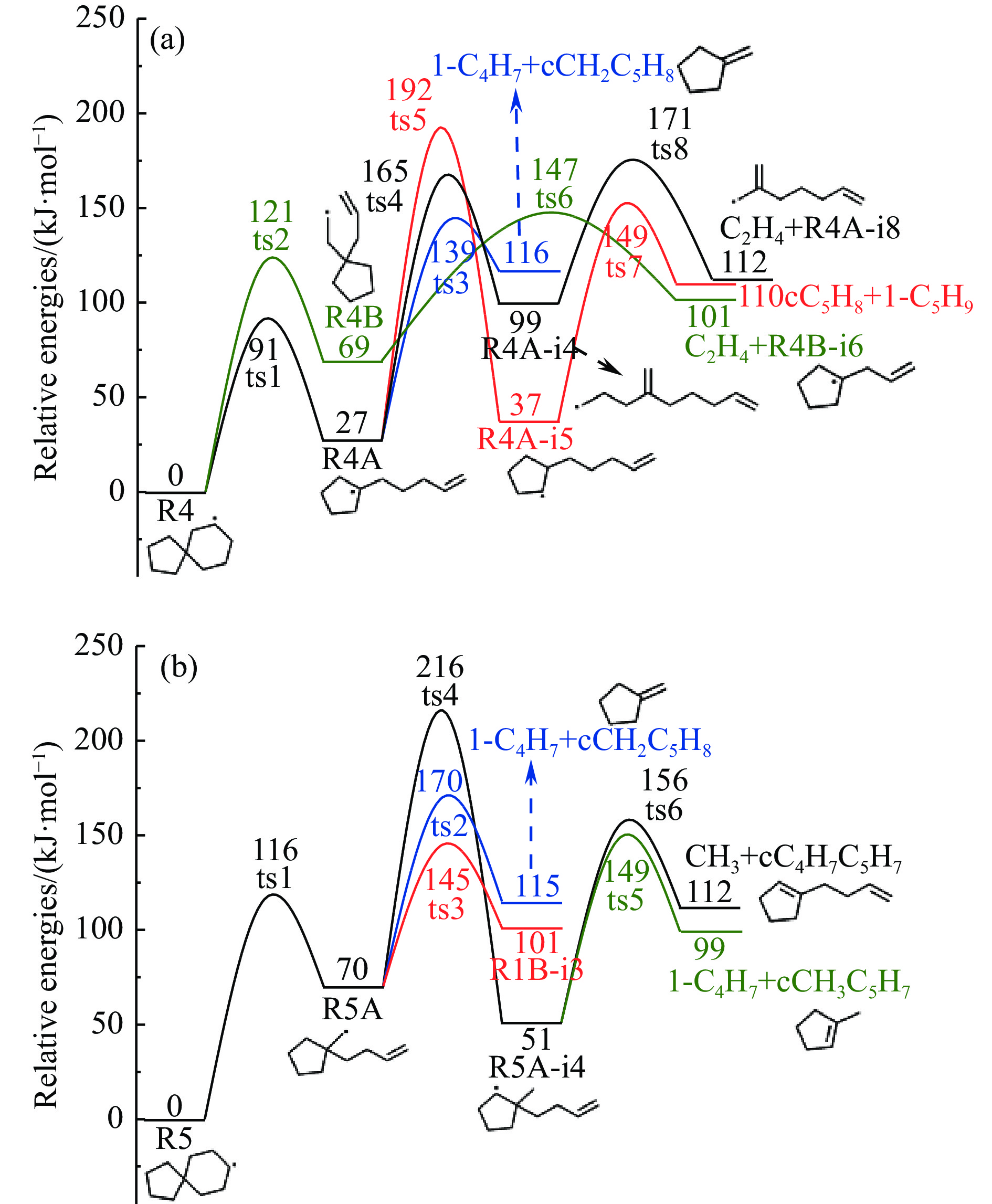
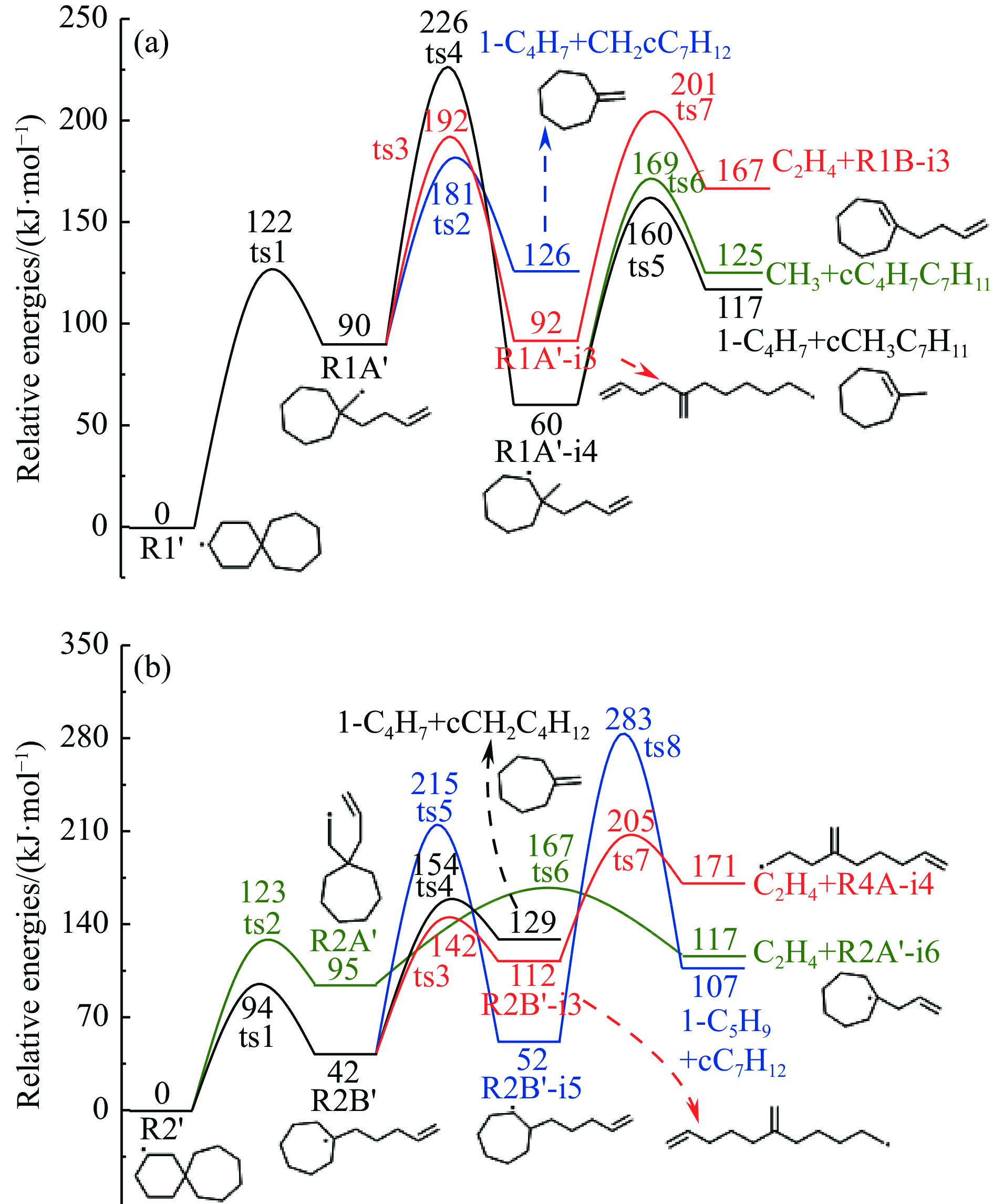
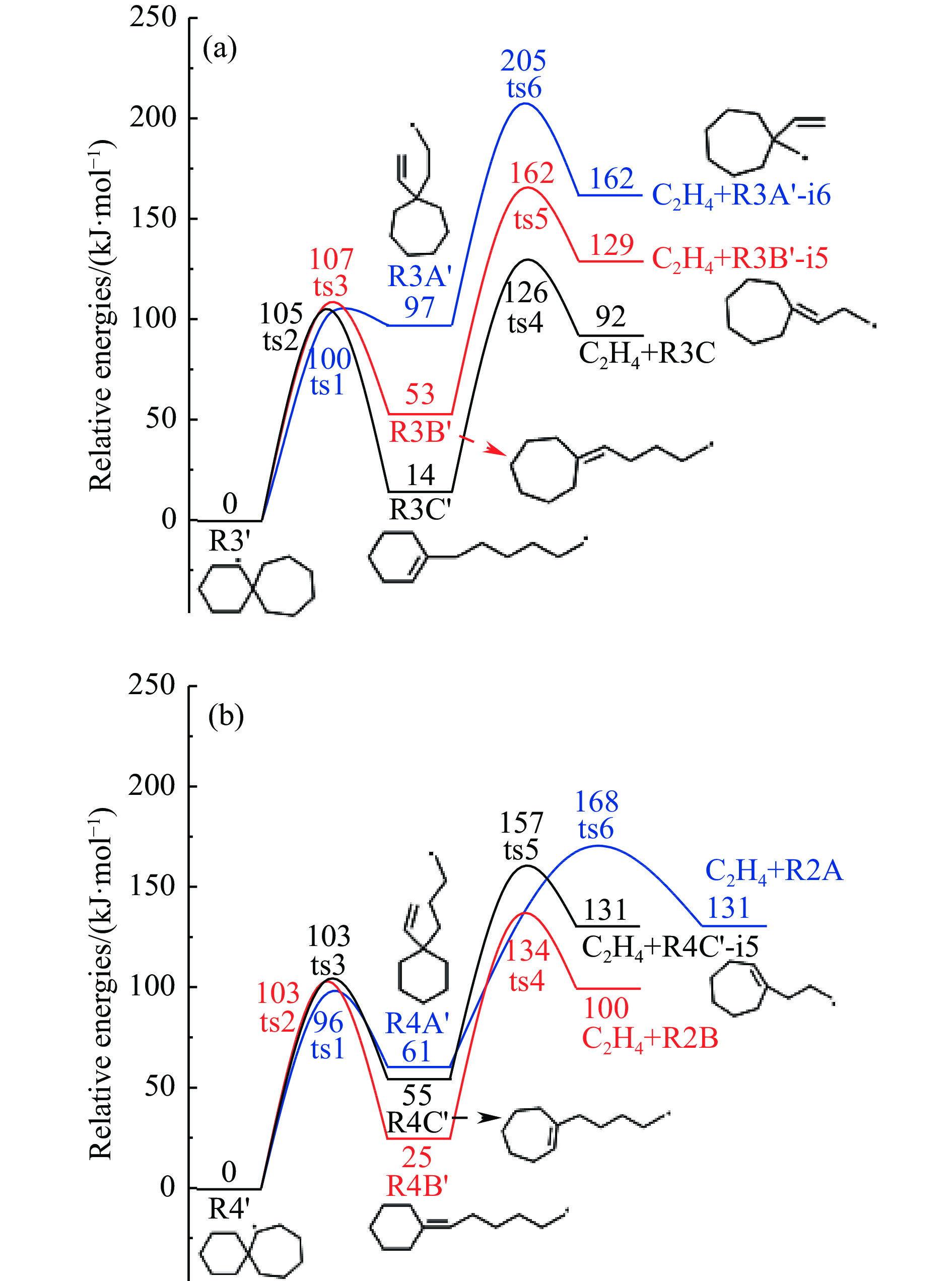

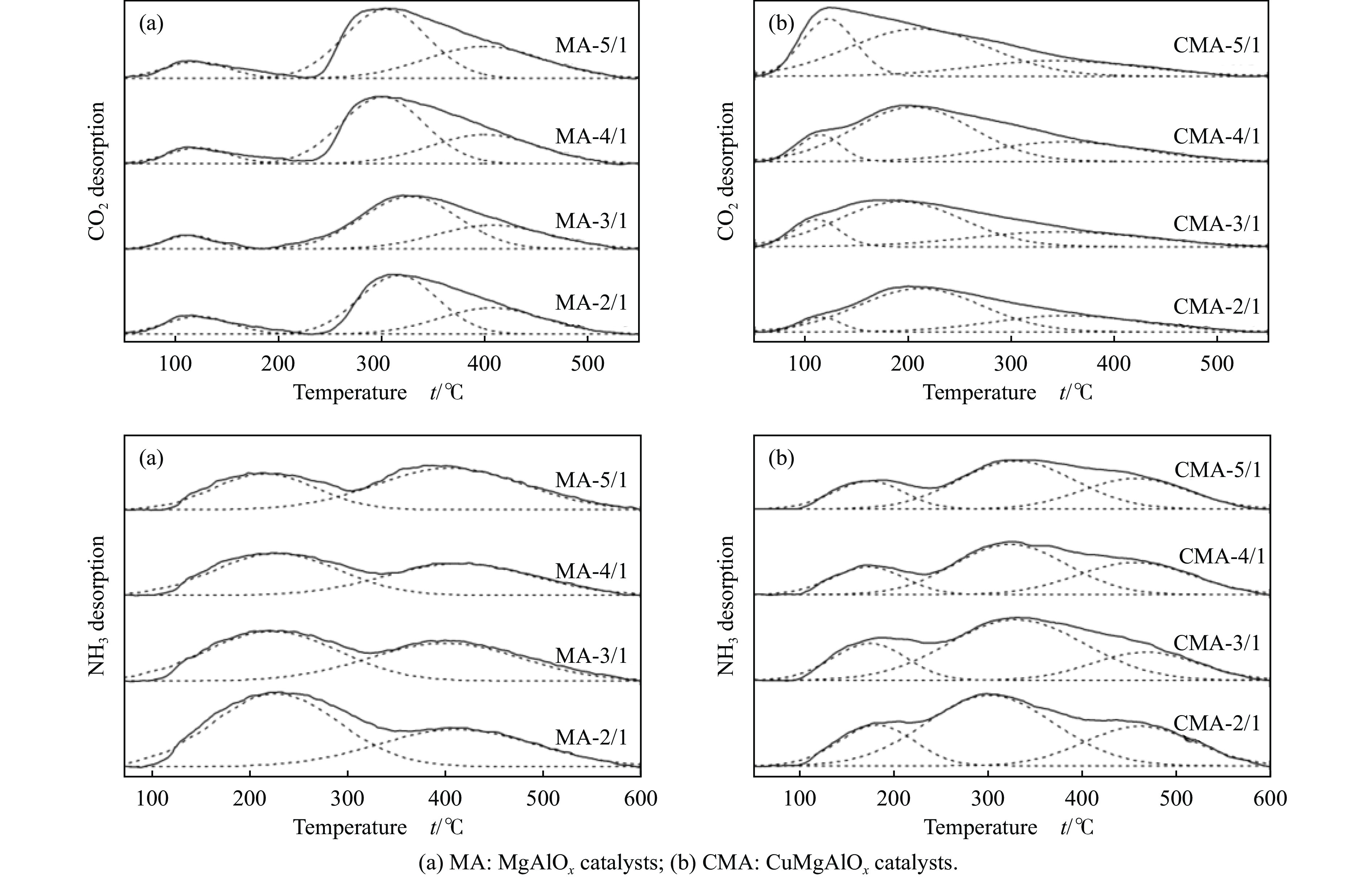
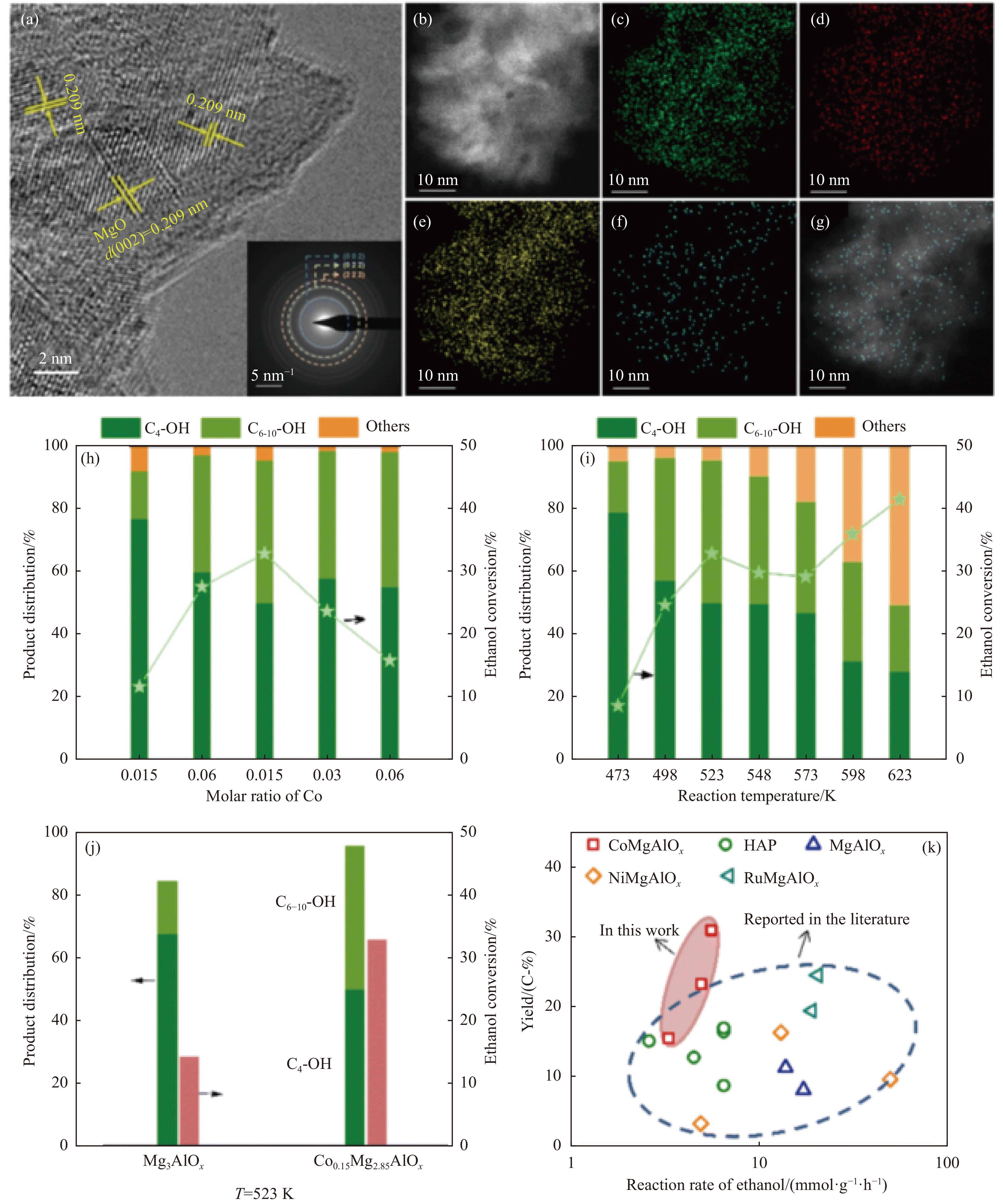
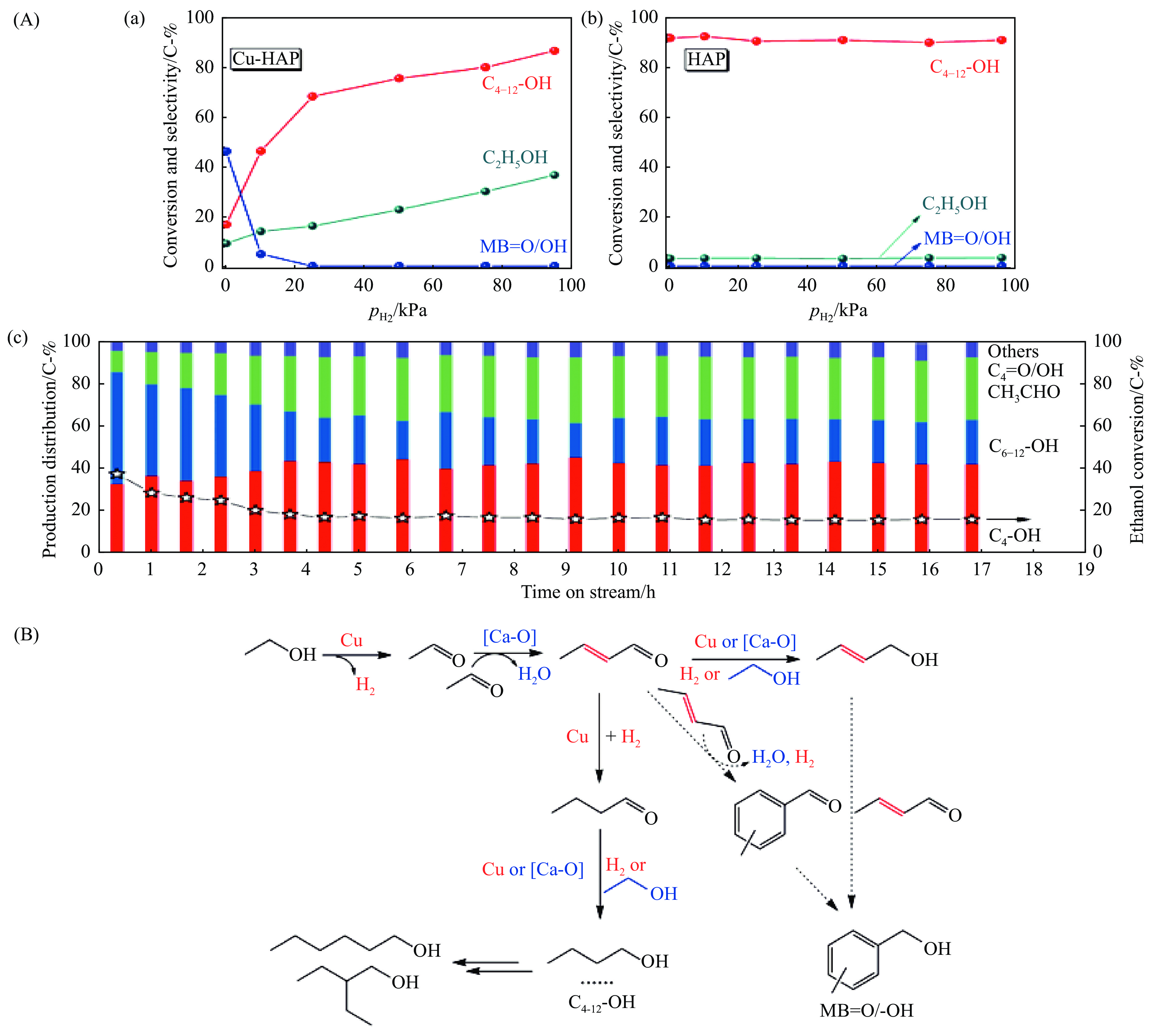
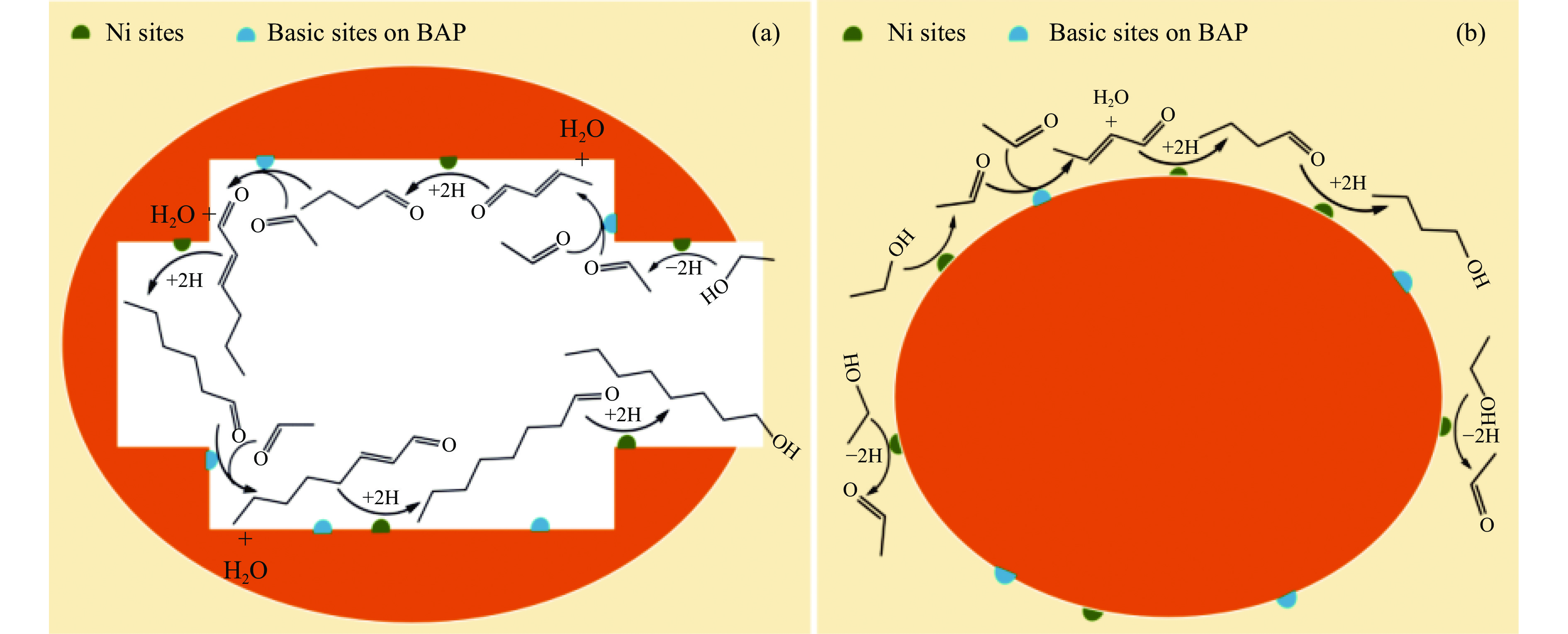
2024, 52(4): 461-480.
doi: 10.19906/j.cnki.JFCT.2023061
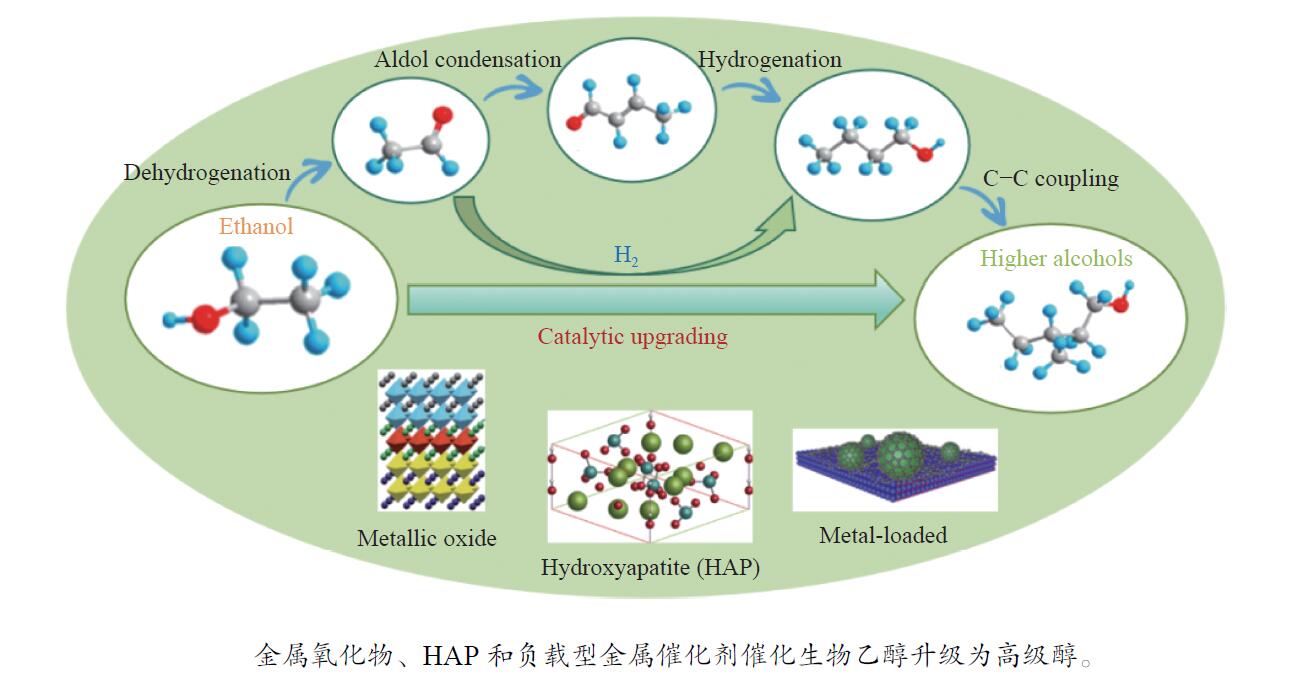
摘要:
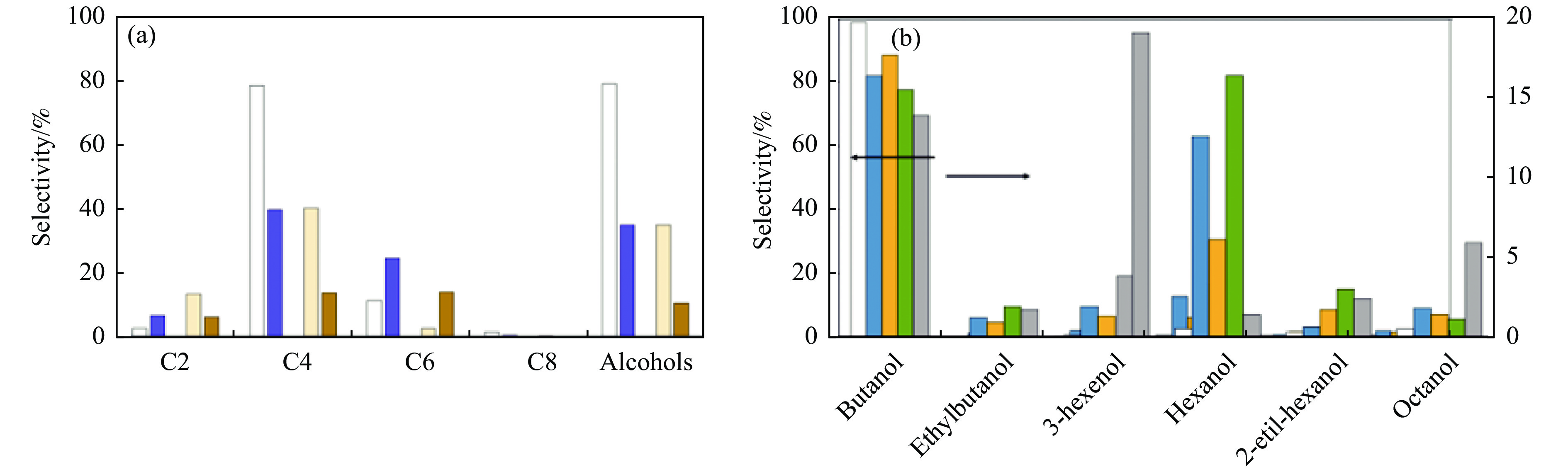
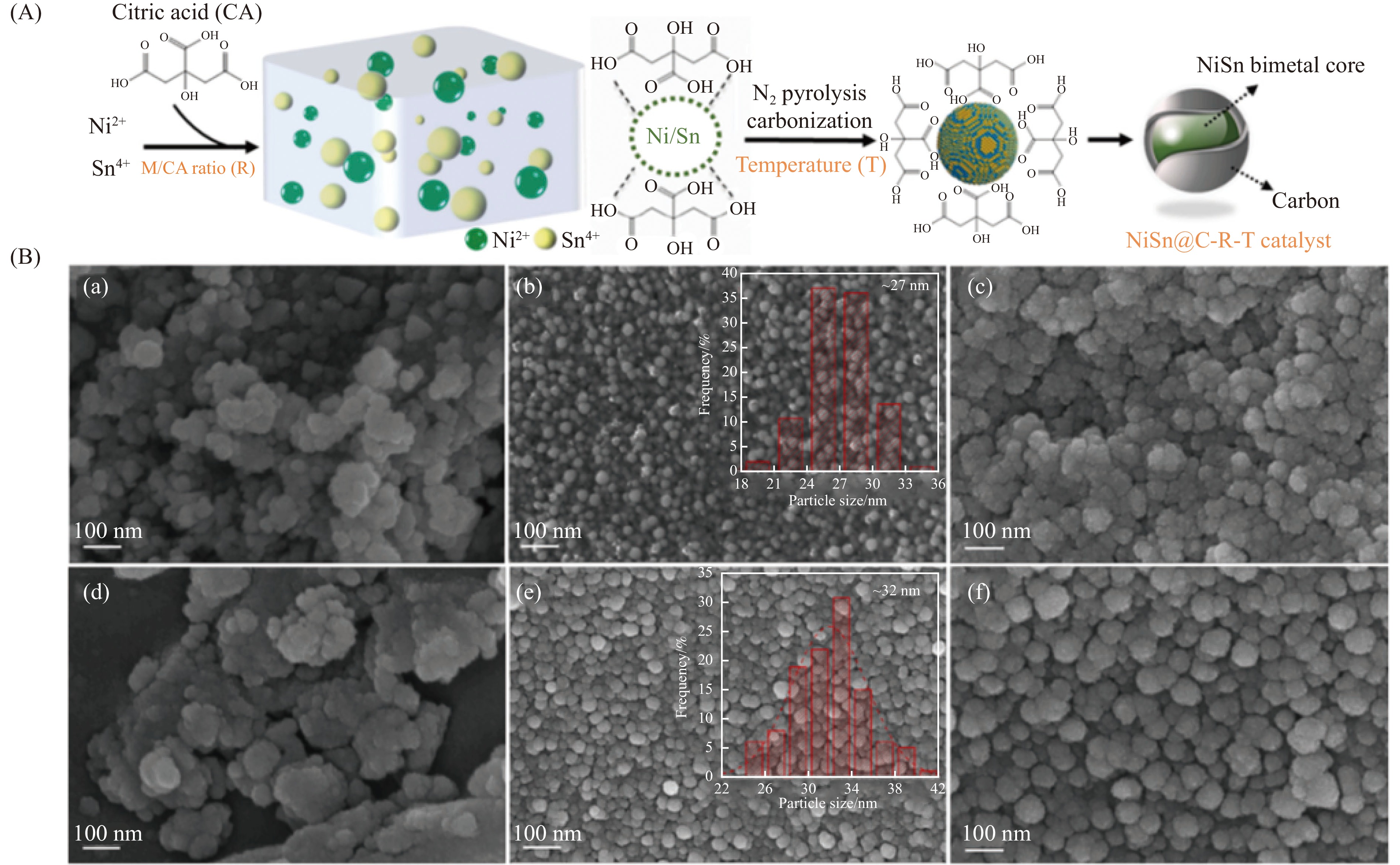
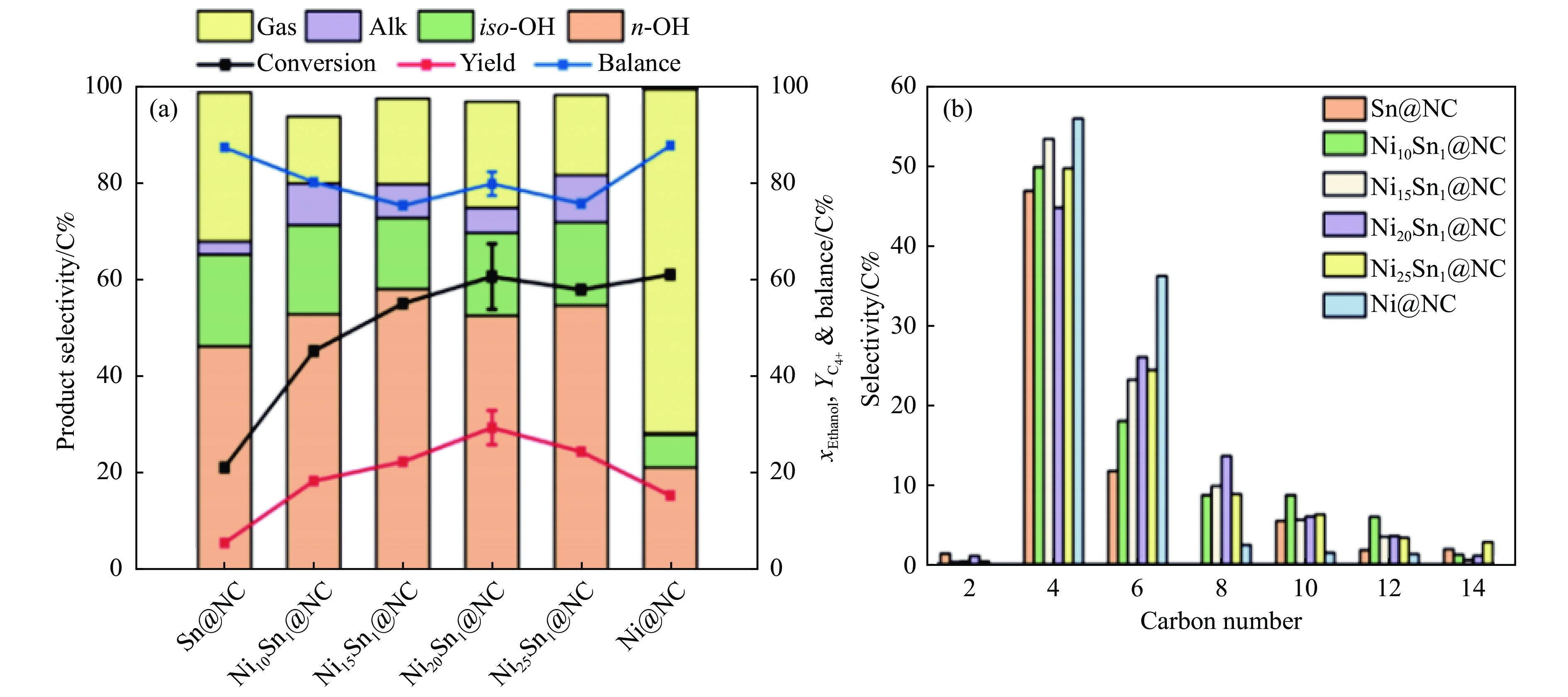
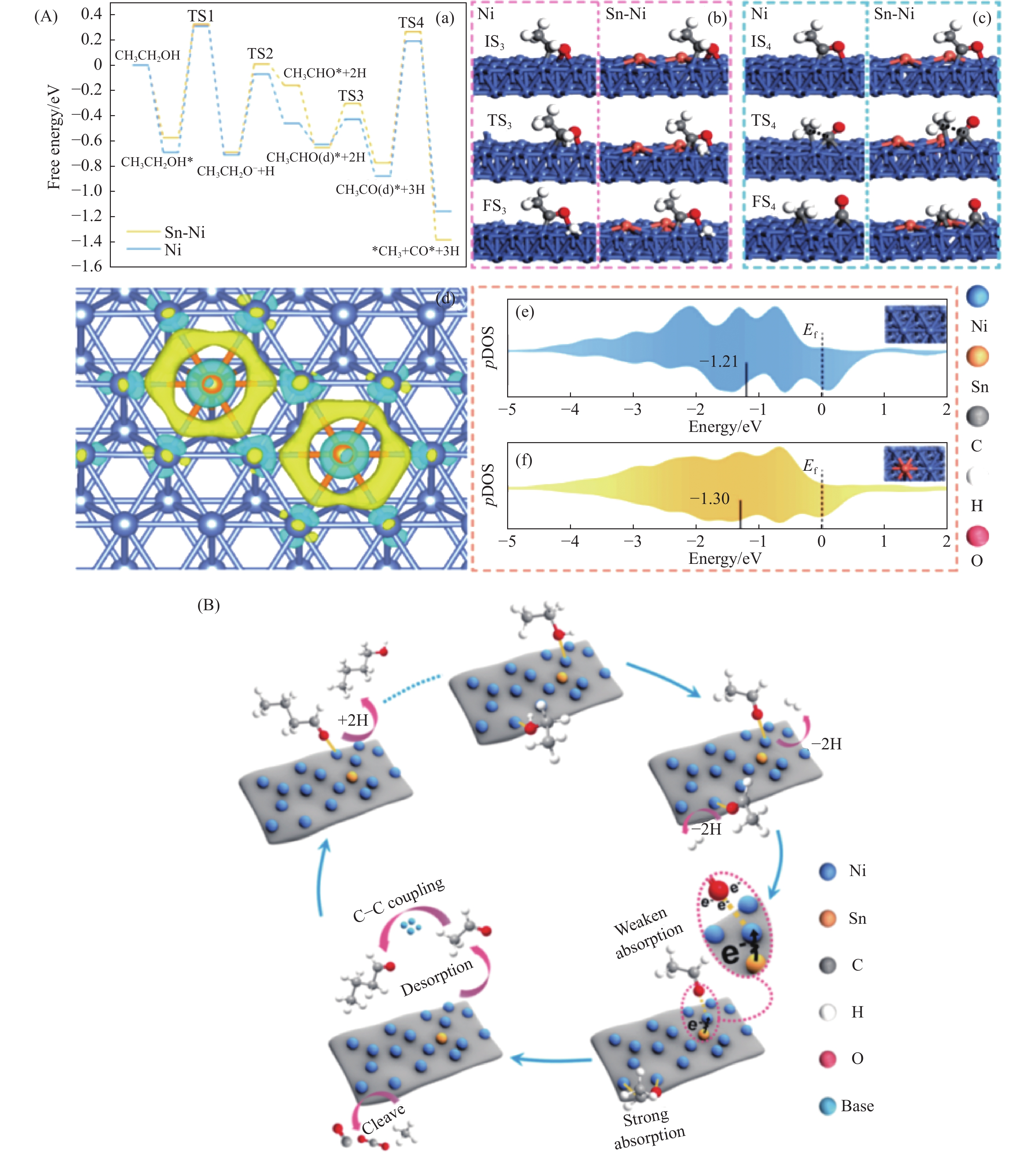
与乙醇相比,高级醇具有高的十六烷值、高能量密度、对发动机部件无腐蚀性、与水不混溶、稳定性好等直接作为燃料或燃料添加剂的优势,将发酵产生的生物乙醇转化为更有价值的高级醇受到了广泛关注。本工作综述了近年来世界各国有关生物乙醇制高级醇的研究进展,包括金属氧化物、羟基磷灰石(HAP)和负载型金属催化剂的研究开发现状,并比较了不同类型催化剂参与下的乙醇转化率和高级醇选择性,结合乙醇经缩合反应制备高级醇的机理进行了讨论,最后对当前生物乙醇制高级醇的挑战以及未来研究趋势进行了总结与展望,指出多功能催化剂的开发是未来研究重点,羟醛缩合是进一步提高生物乙醇制高级醇转化率与选择性的有效策略。
与乙醇相比,高级醇具有高的十六烷值、高能量密度、对发动机部件无腐蚀性、与水不混溶、稳定性好等直接作为燃料或燃料添加剂的优势,将发酵产生的生物乙醇转化为更有价值的高级醇受到了广泛关注。本工作综述了近年来世界各国有关生物乙醇制高级醇的研究进展,包括金属氧化物、羟基磷灰石(HAP)和负载型金属催化剂的研究开发现状,并比较了不同类型催化剂参与下的乙醇转化率和高级醇选择性,结合乙醇经缩合反应制备高级醇的机理进行了讨论,最后对当前生物乙醇制高级醇的挑战以及未来研究趋势进行了总结与展望,指出多功能催化剂的开发是未来研究重点,羟醛缩合是进一步提高生物乙醇制高级醇转化率与选择性的有效策略。




2024, 52(4): 481-495.
doi: 10.1016/S1872-5813(23)60401-3
摘要:
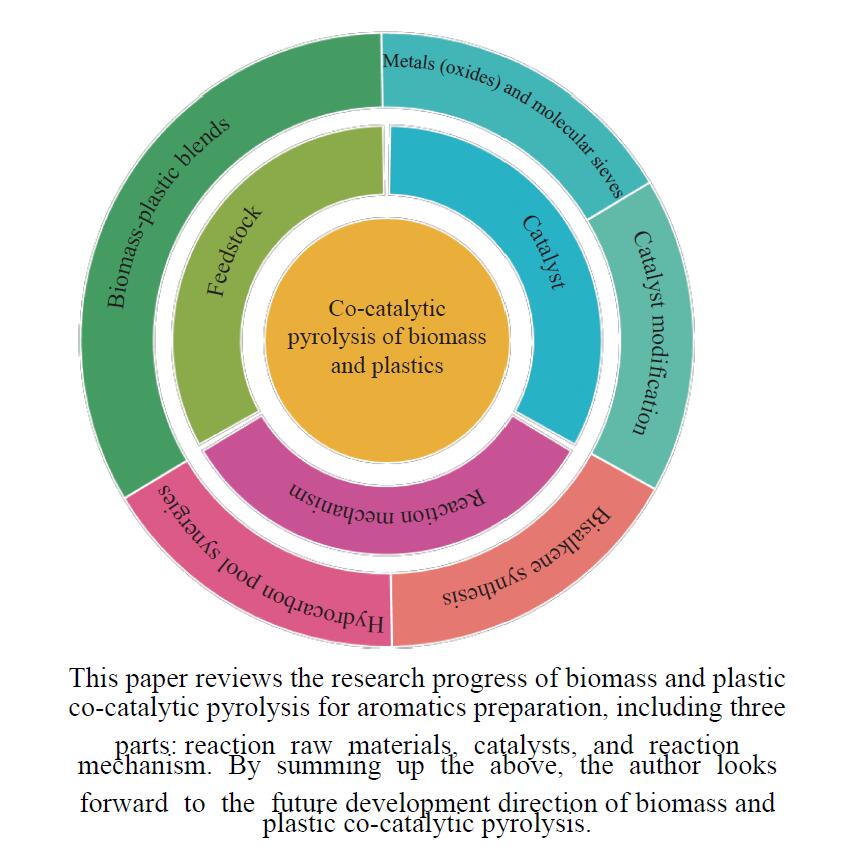
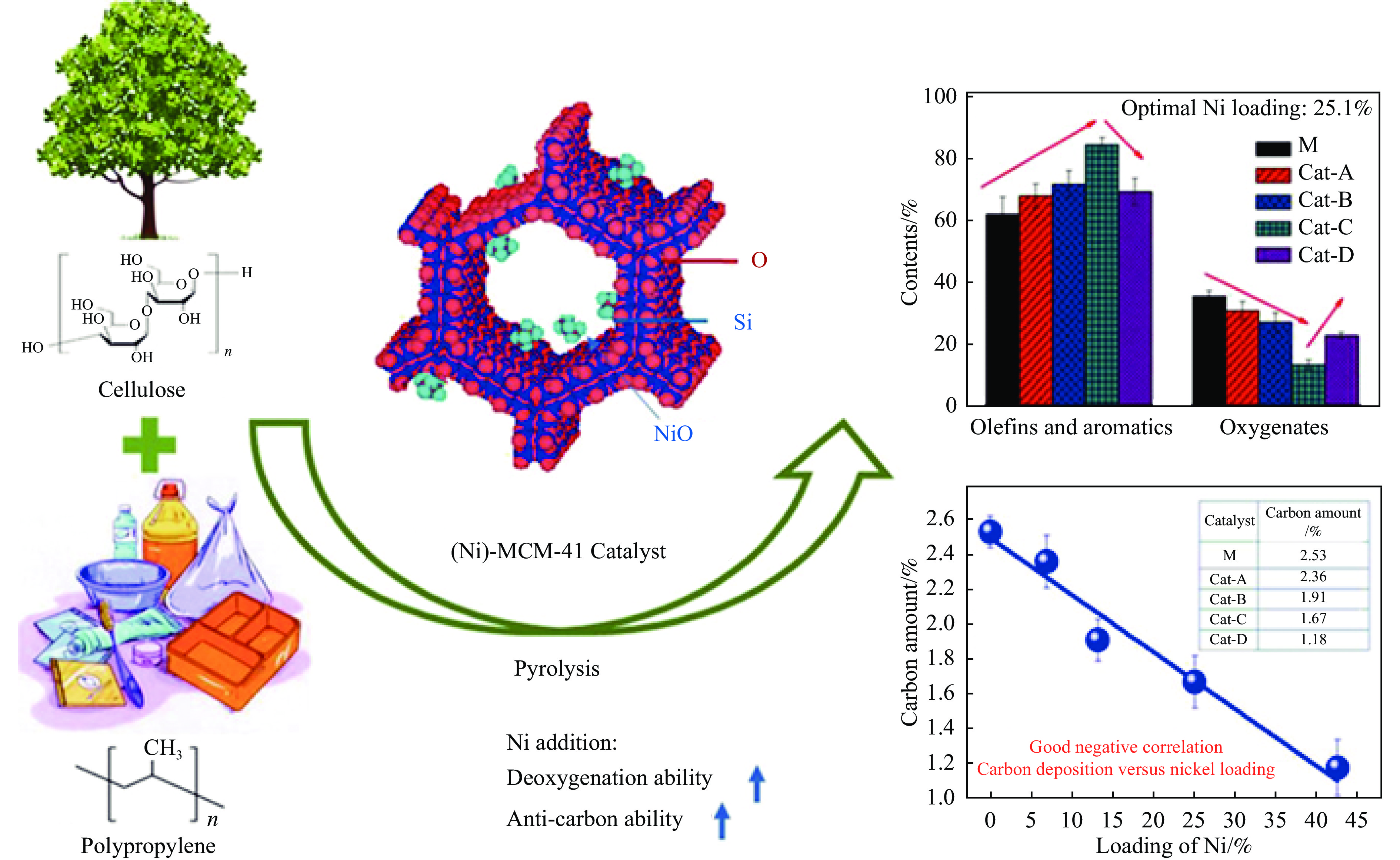
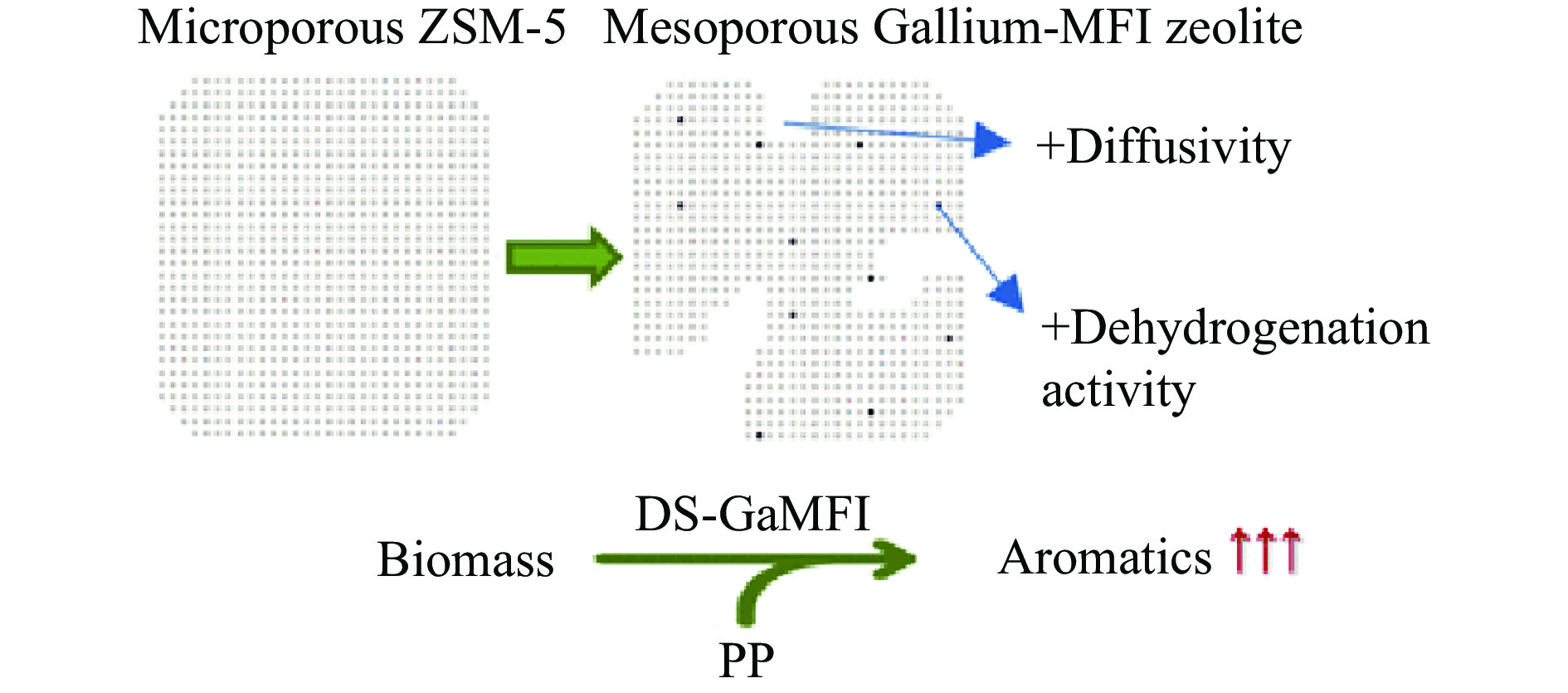
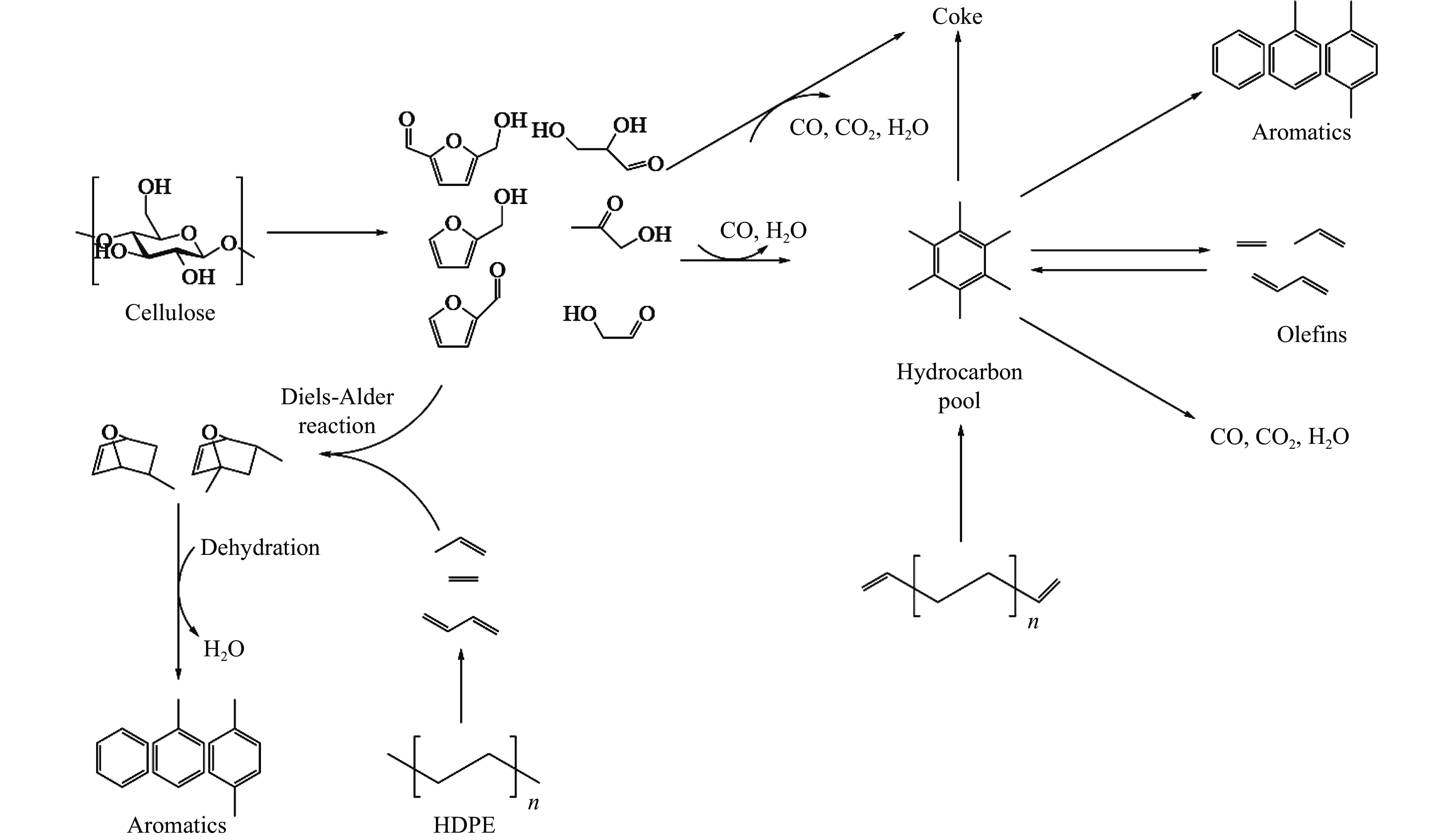
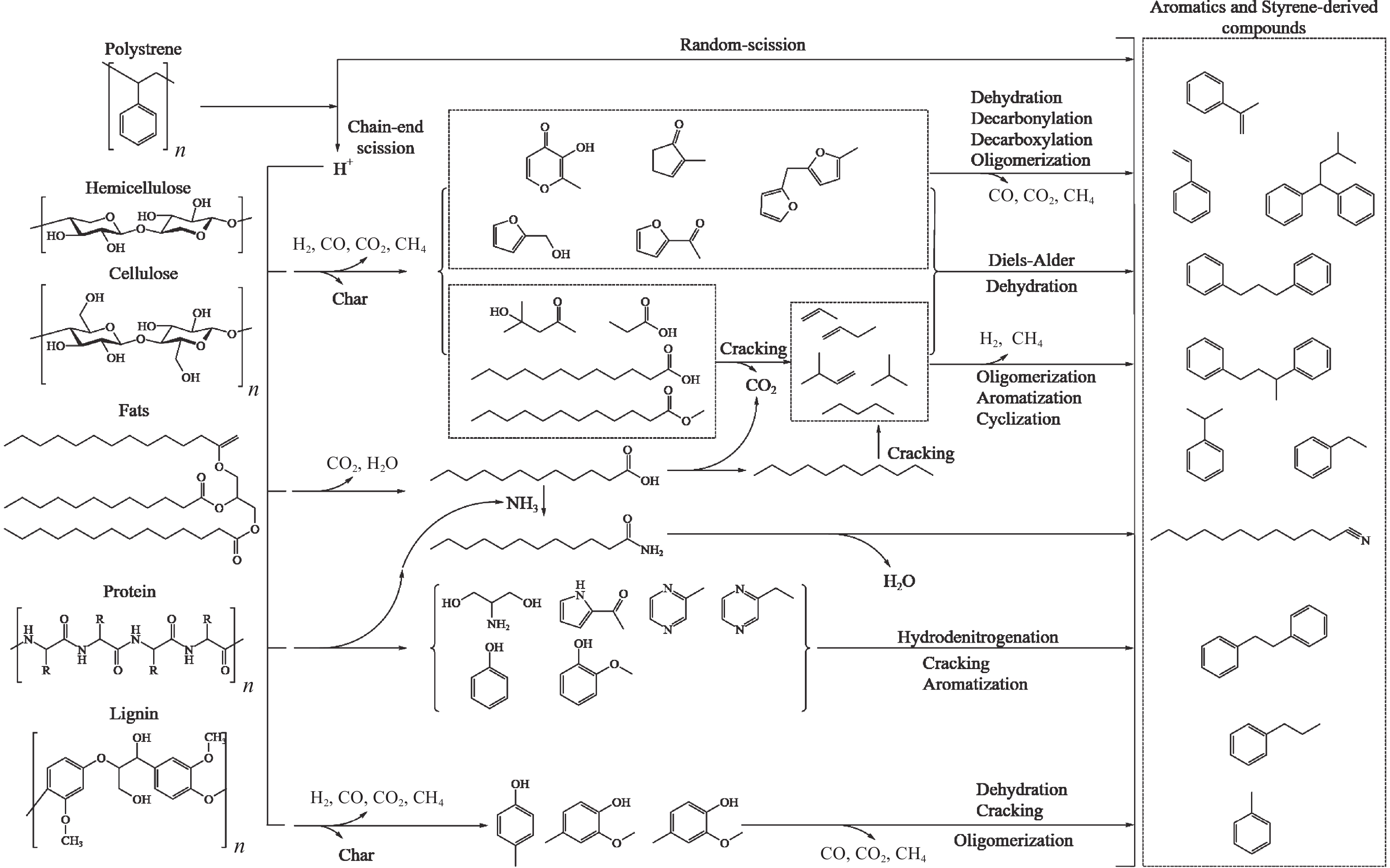
芳烃,尤其苯、甲苯、二甲苯(BTX)等单环芳烃,是化工行业重要的基础原料,主要来源于化石燃料的催化重整与热裂解。生物质与塑料共催化热解制芳烃具有高效、环保、低成本、高选择性等优点,可以解决因生物质富氧、贫氢的特点造成热解产物氧含量高、芳烃收率和选择性低等问题。本工作主要综述了生物质与塑料共催化热解制备芳烃化合物的研究进展,介绍了共催化热解反应原料类别,重点论述了共催化热解反应催化剂,总结归纳了共催化热解双烯合成、烃池协同等反应机理。展望了生物质与塑料共催化热解未来的研究重心与发展方向,即通过研制高活性、高稳定性的改性分子筛催化剂来提高芳烃产率。
芳烃,尤其苯、甲苯、二甲苯(BTX)等单环芳烃,是化工行业重要的基础原料,主要来源于化石燃料的催化重整与热裂解。生物质与塑料共催化热解制芳烃具有高效、环保、低成本、高选择性等优点,可以解决因生物质富氧、贫氢的特点造成热解产物氧含量高、芳烃收率和选择性低等问题。本工作主要综述了生物质与塑料共催化热解制备芳烃化合物的研究进展,介绍了共催化热解反应原料类别,重点论述了共催化热解反应催化剂,总结归纳了共催化热解双烯合成、烃池协同等反应机理。展望了生物质与塑料共催化热解未来的研究重心与发展方向,即通过研制高活性、高稳定性的改性分子筛催化剂来提高芳烃产率。

2024, 52(4): 496-511.
doi: 10.1016/S1872-5813(23)60404-9
摘要:
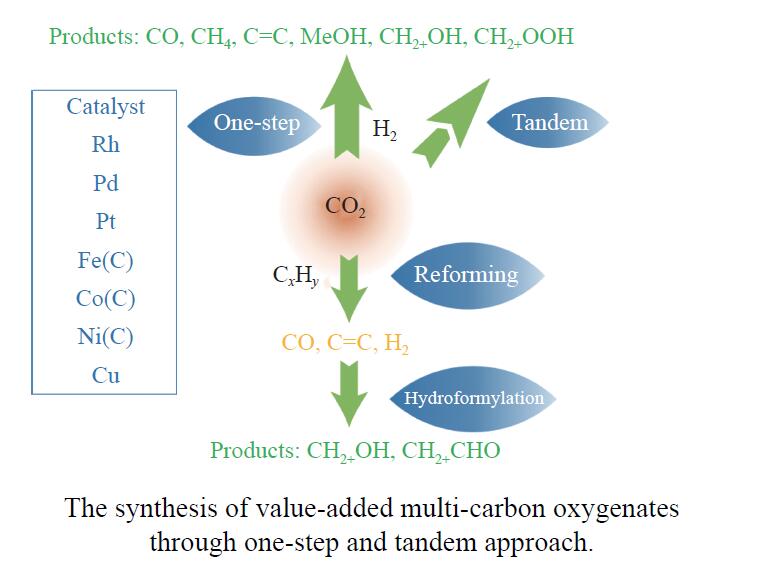
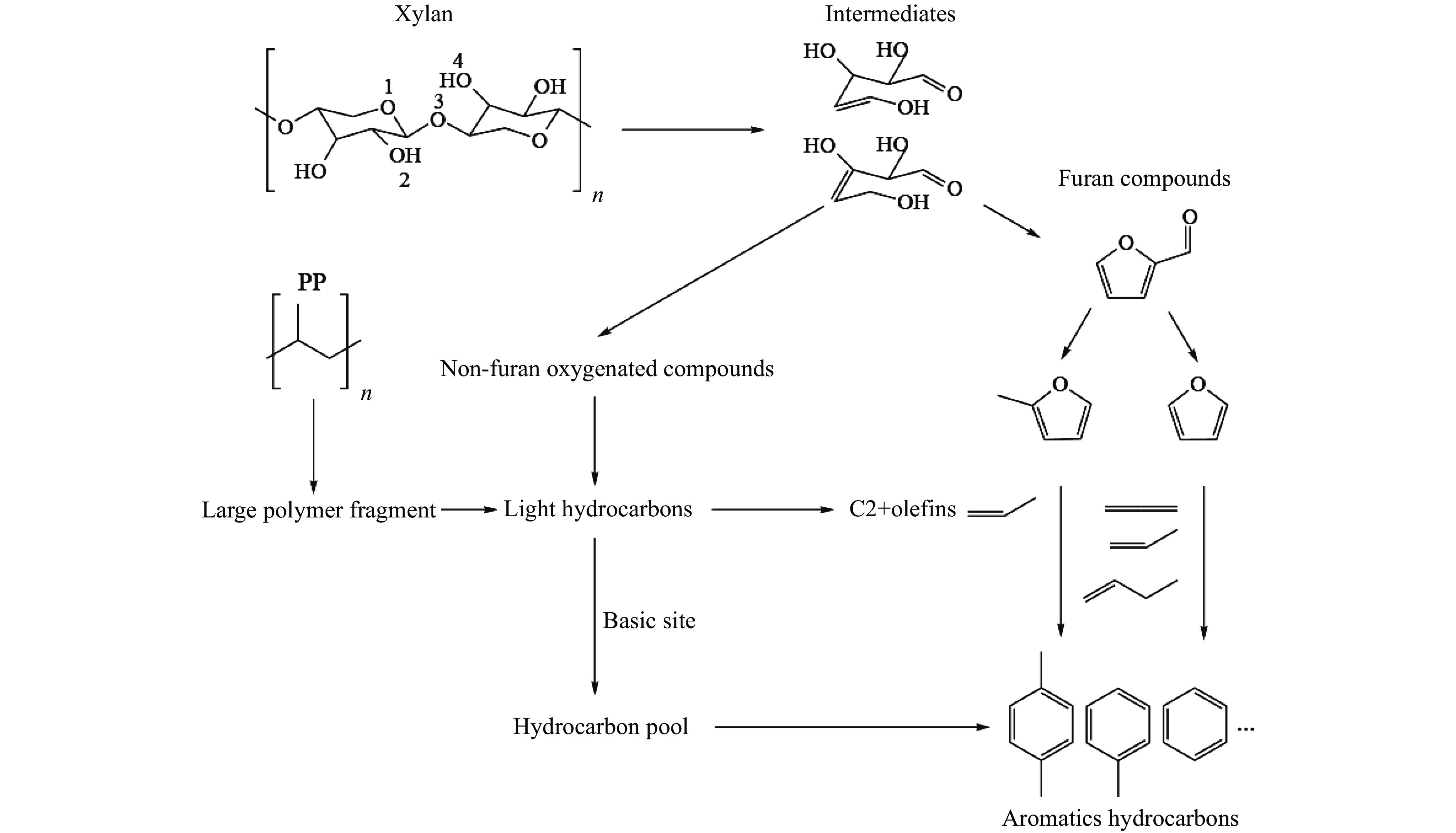
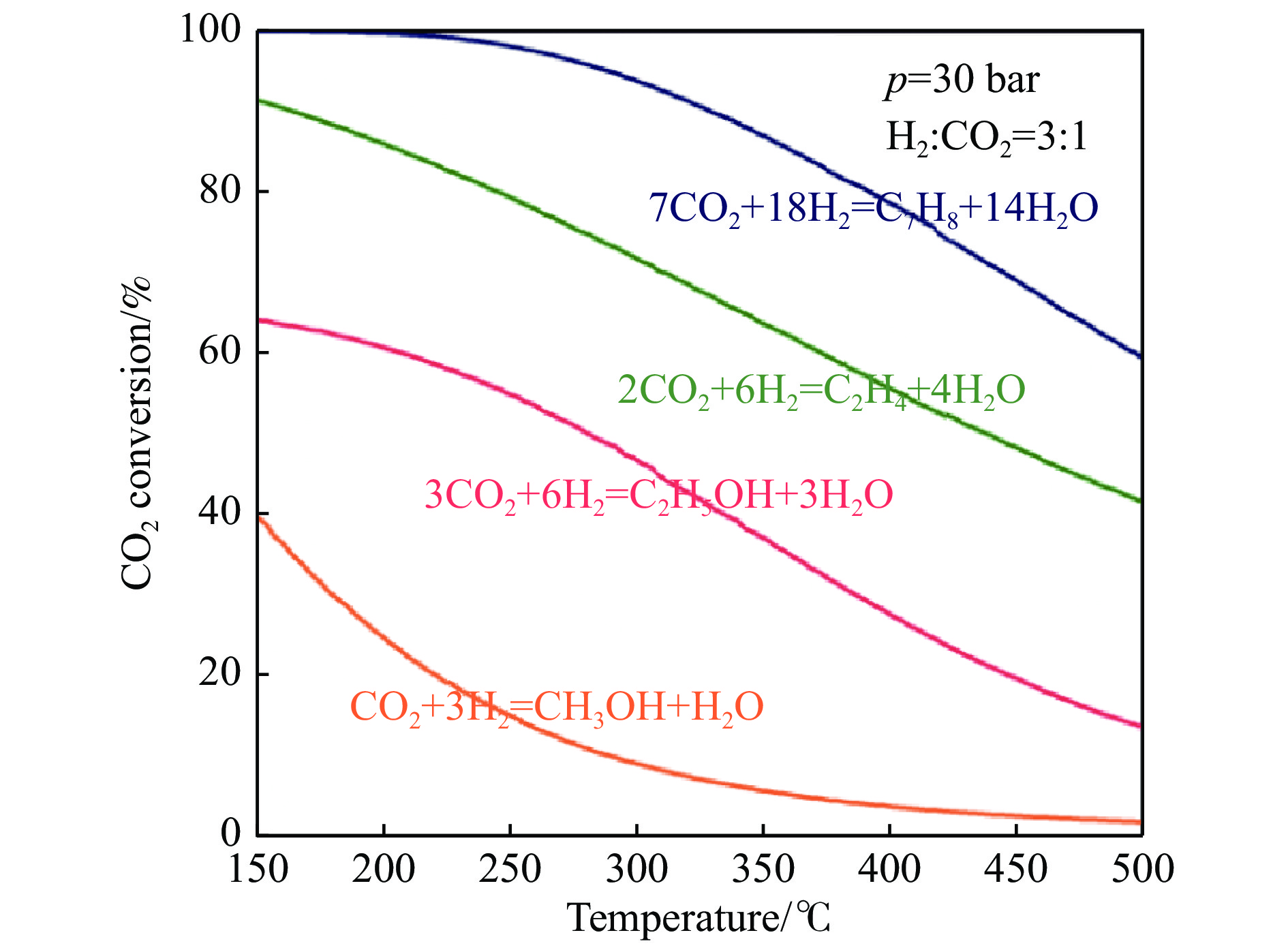
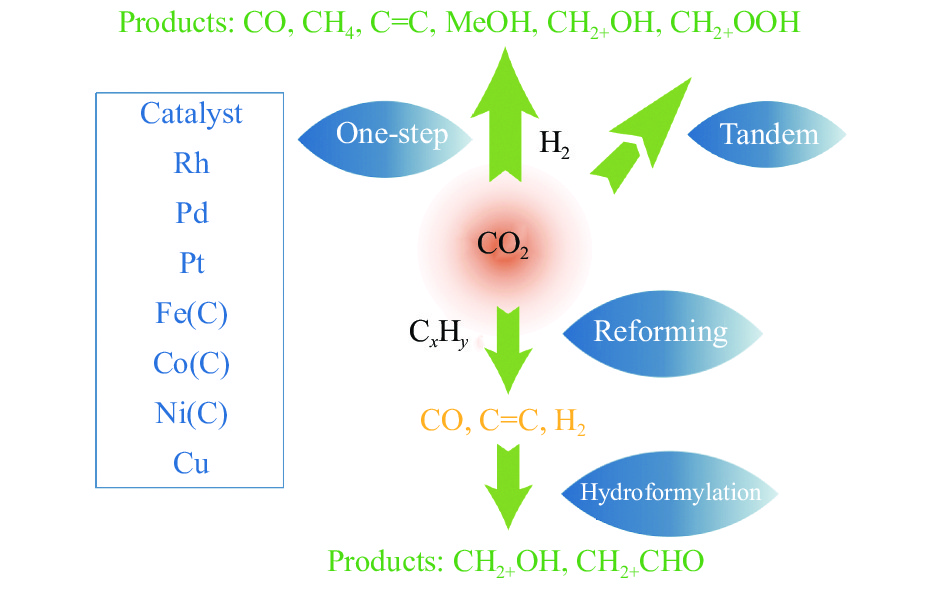
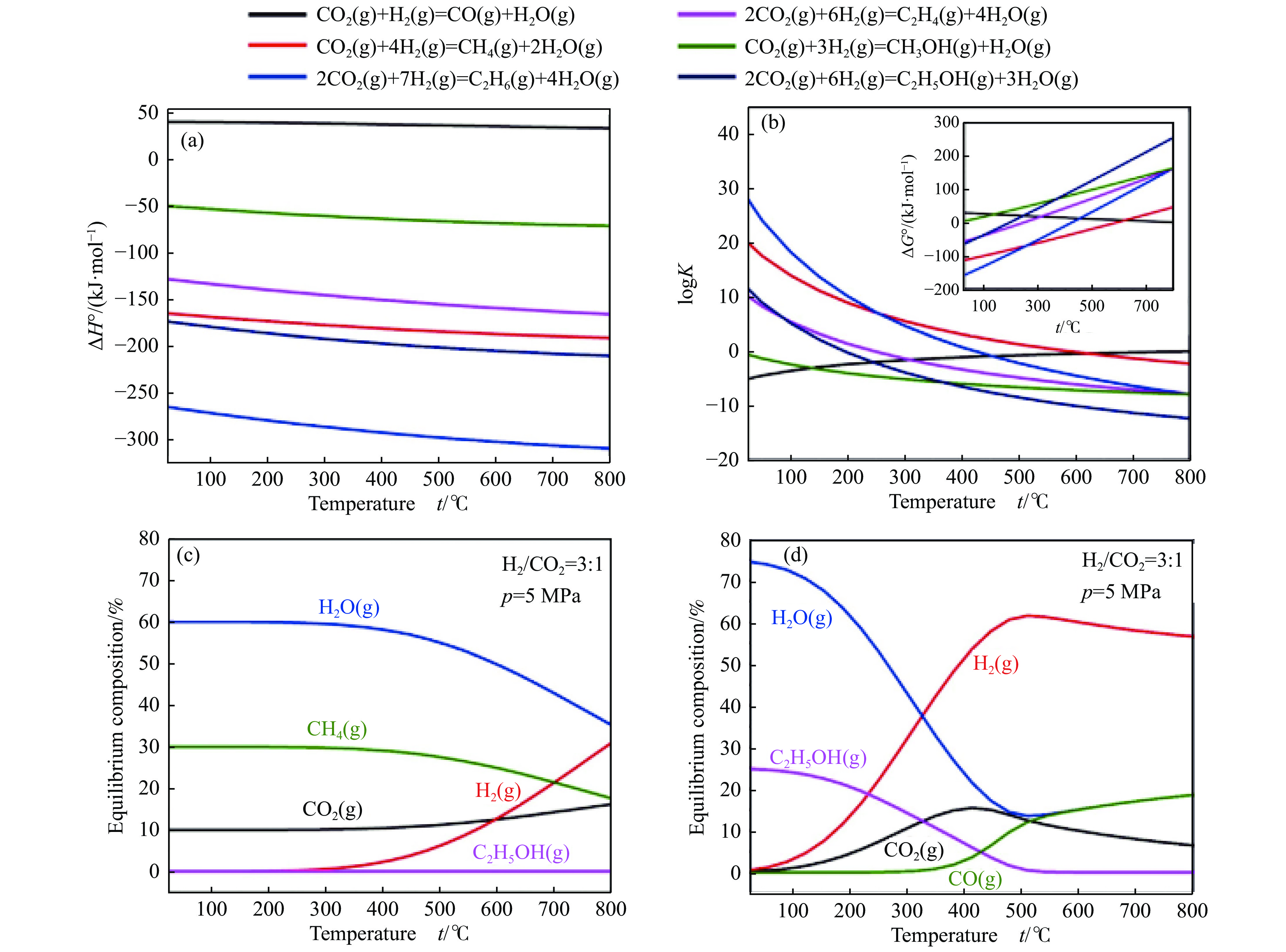
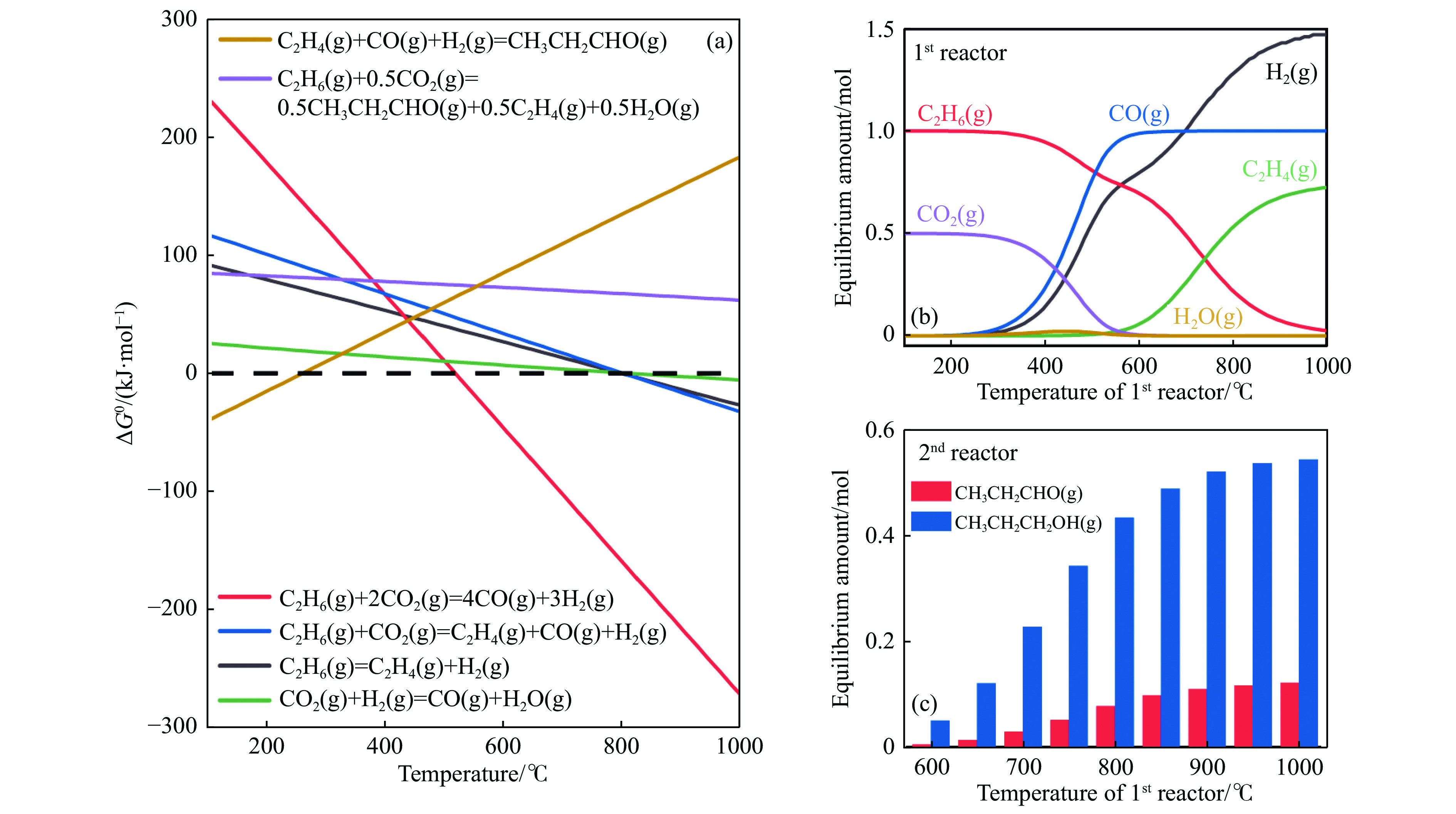
将温室气体CO2通过化学反应路径制备高附加值多碳含氧化合物如乙醇、乙酸、丙醛、丙酸、丁醇等具有挑战性。由于C−C偶联反应的复杂性和成键的不可控性,导致合成多碳高值含氧化合物困难。本工作总结了近期在连续流固定床条件下CO2催化合成高附加值多碳含氧化合物的研究进展。首先归纳了CO2加氢路径下可能的反应机理;其次总结了CO2直接加氢(一步法、串联法)、CO2与轻烃重整、CO2氢甲酰化等不同反应路径下具有潜力的催化剂,包括金属碳化物、碱金属修饰的Cu、Fe、Co、Rh等单金属或二元金属制备多碳高值含氧化合物的特点,并进一步阐述了不同催化剂上的作用机制。最后对目前存在的问题和未来可能的解决方案进行了讨论和展望。
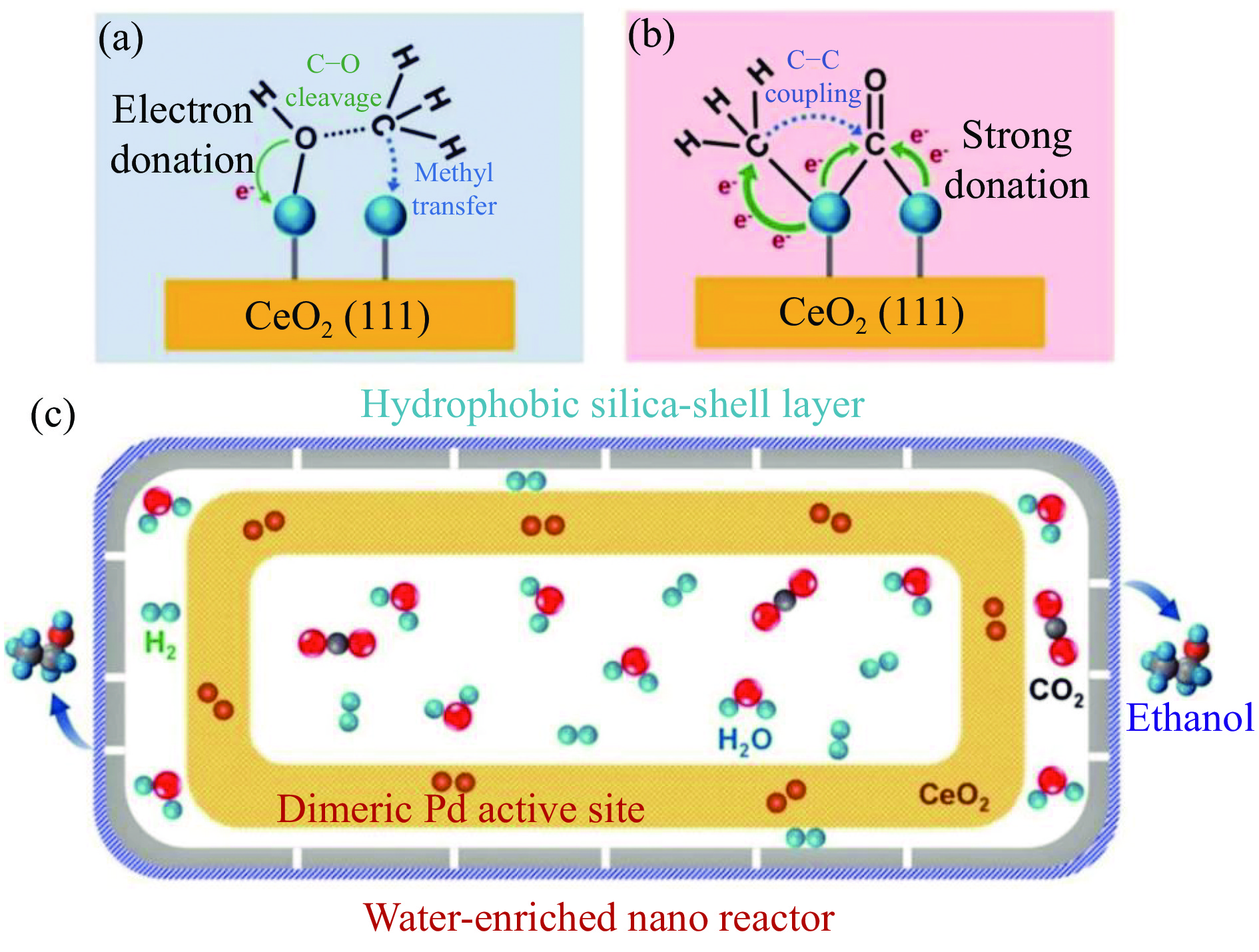
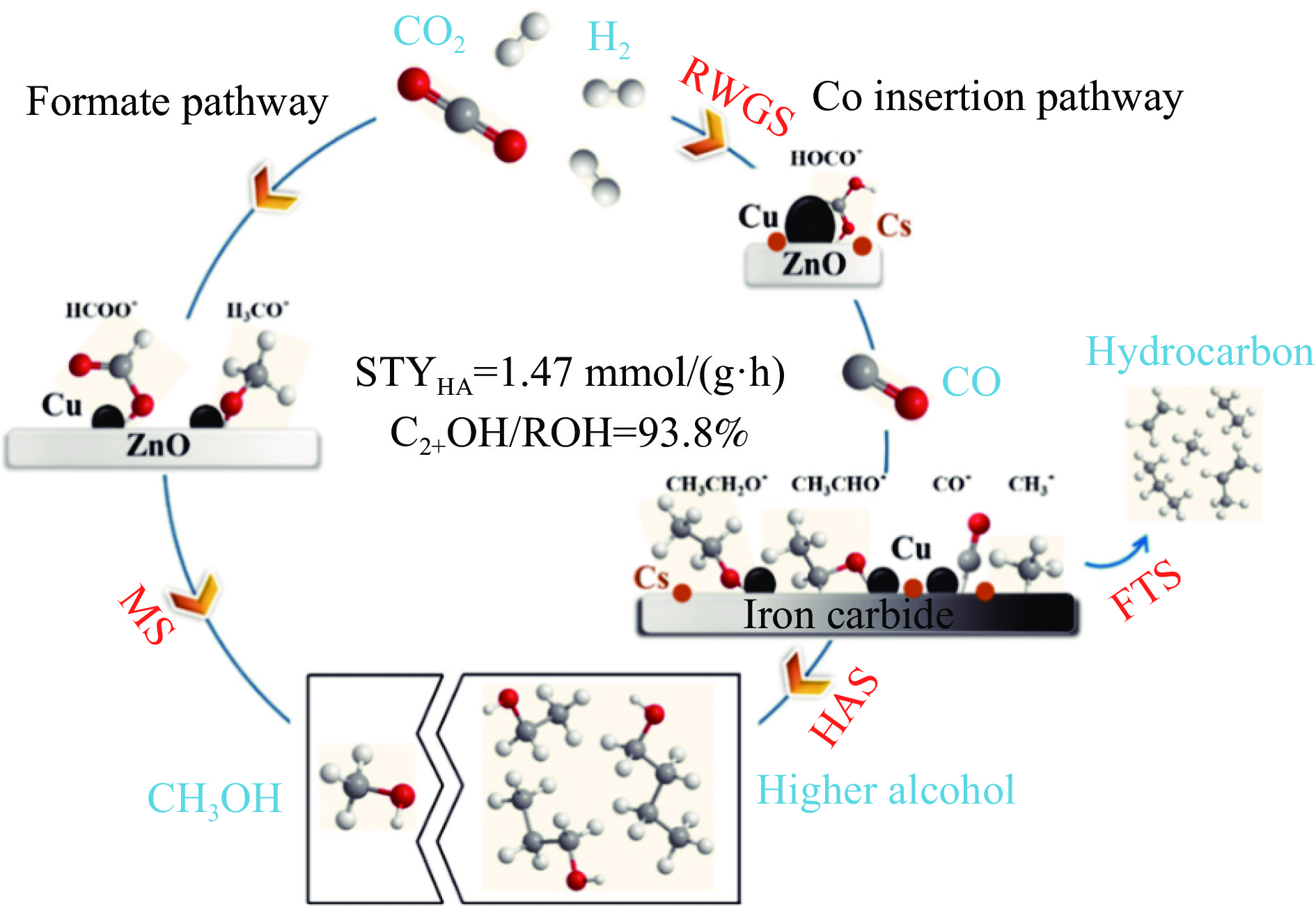
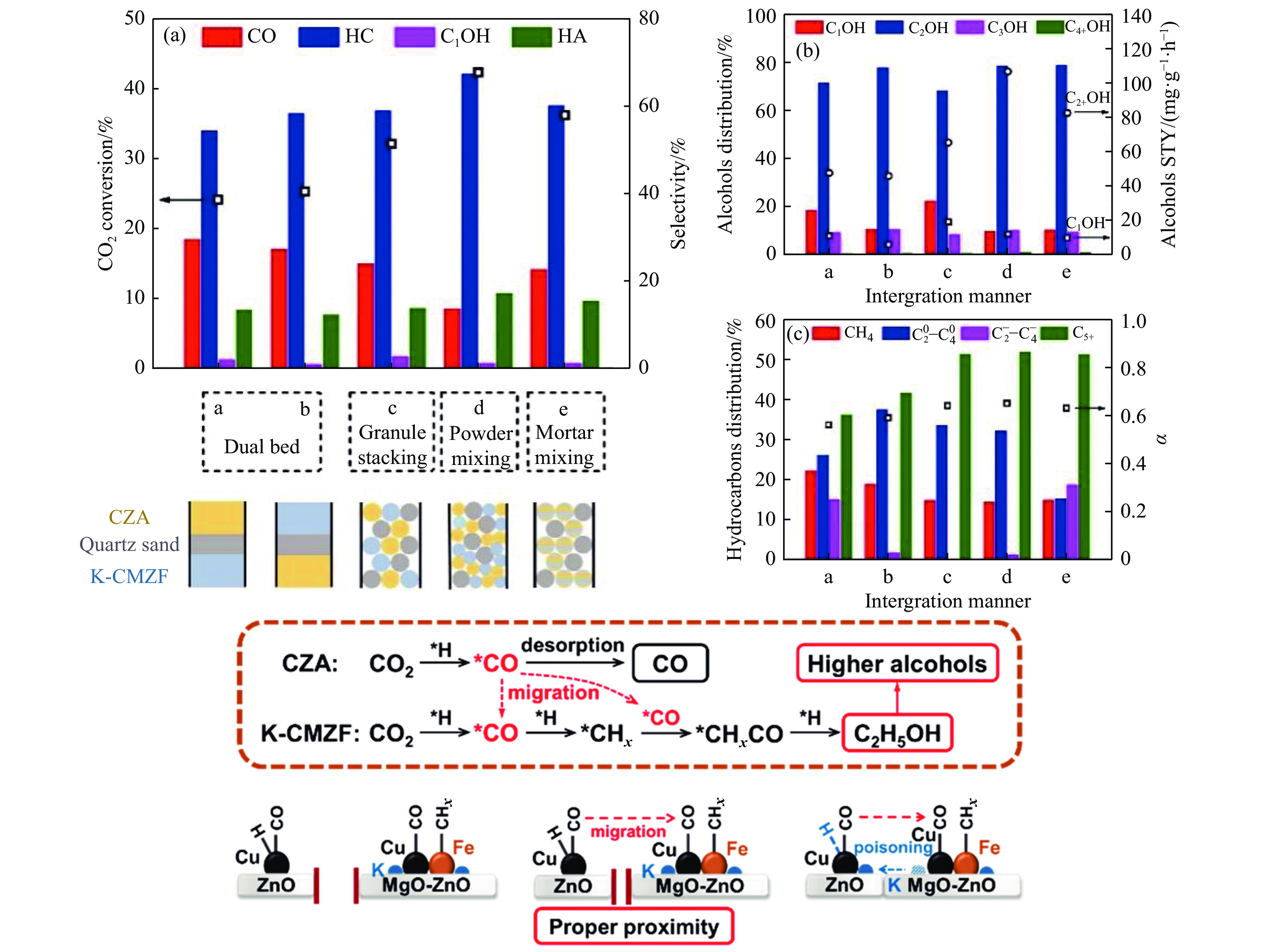
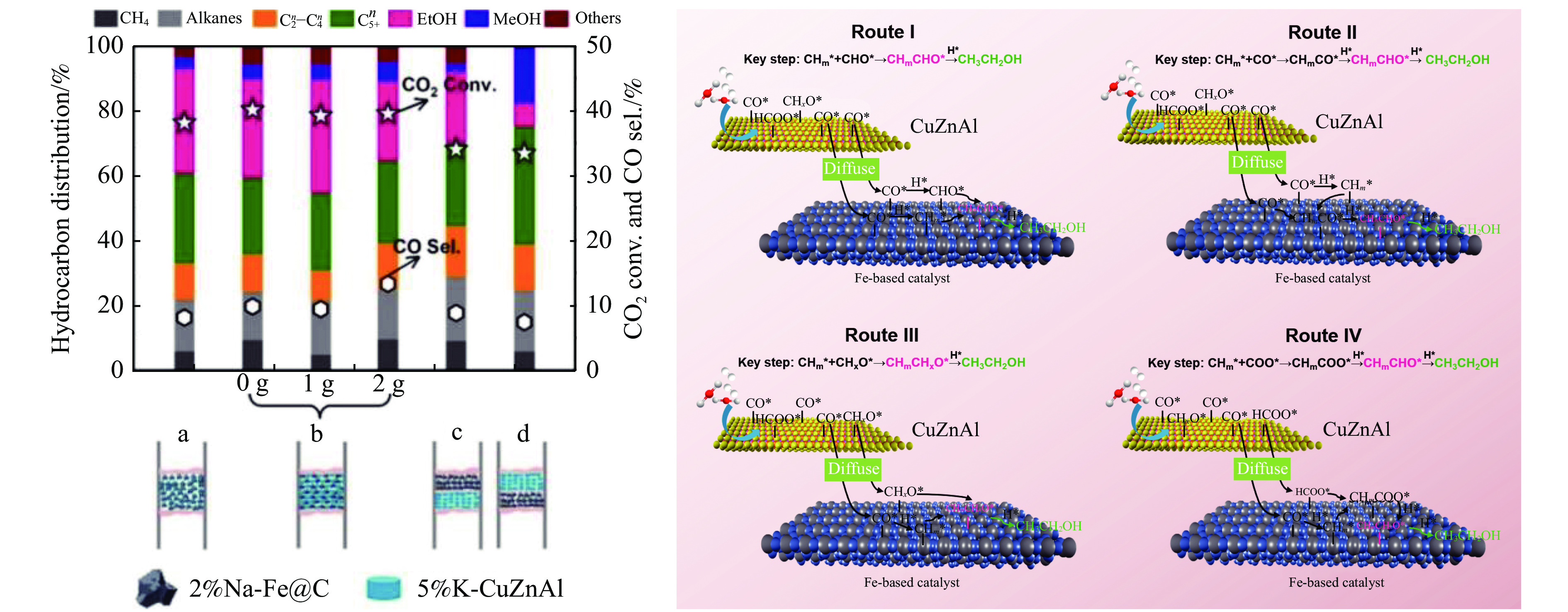
将温室气体CO2通过化学反应路径制备高附加值多碳含氧化合物如乙醇、乙酸、丙醛、丙酸、丁醇等具有挑战性。由于C−C偶联反应的复杂性和成键的不可控性,导致合成多碳高值含氧化合物困难。本工作总结了近期在连续流固定床条件下CO2催化合成高附加值多碳含氧化合物的研究进展。首先归纳了CO2加氢路径下可能的反应机理;其次总结了CO2直接加氢(一步法、串联法)、CO2与轻烃重整、CO2氢甲酰化等不同反应路径下具有潜力的催化剂,包括金属碳化物、碱金属修饰的Cu、Fe、Co、Rh等单金属或二元金属制备多碳高值含氧化合物的特点,并进一步阐述了不同催化剂上的作用机制。最后对目前存在的问题和未来可能的解决方案进行了讨论和展望。


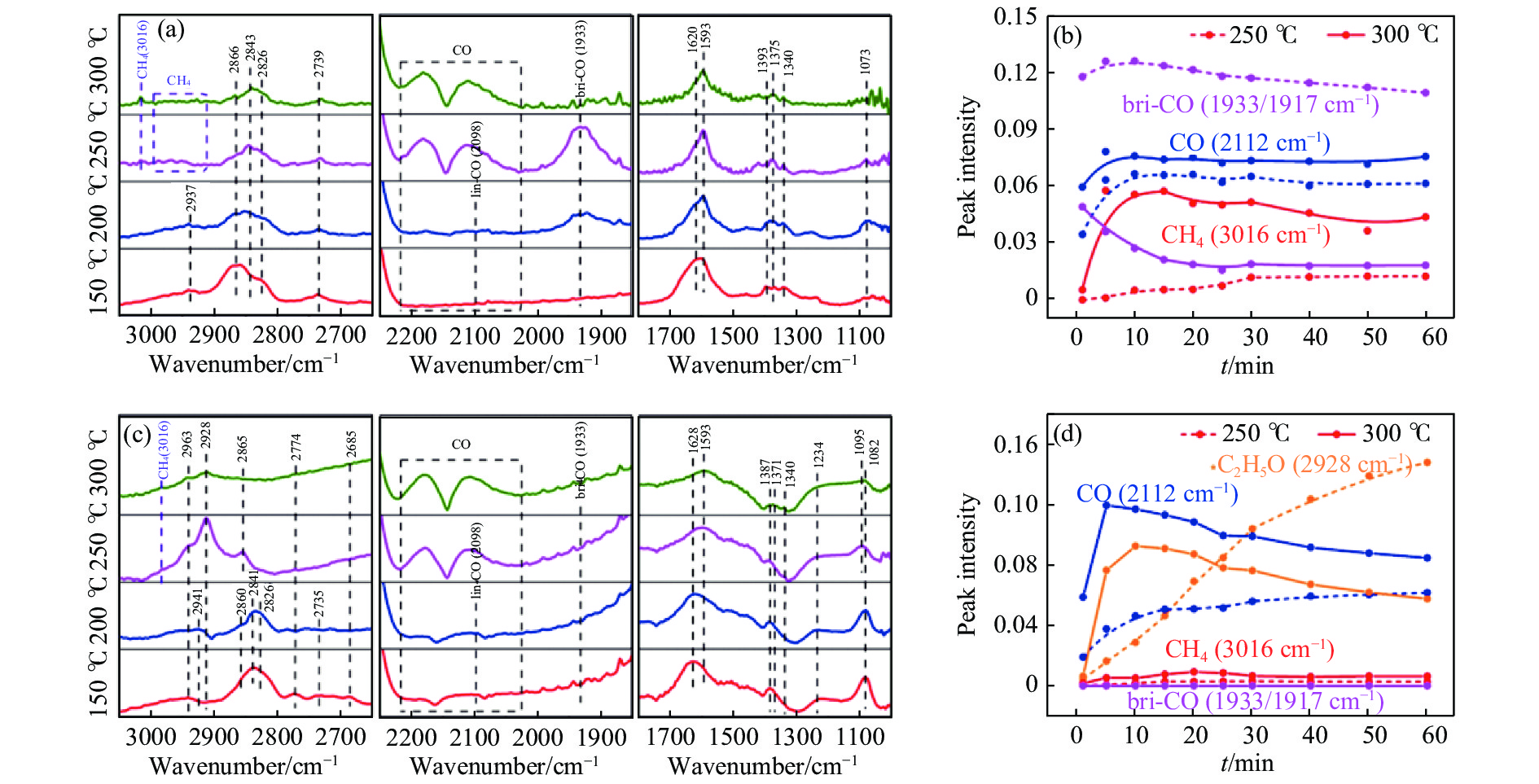
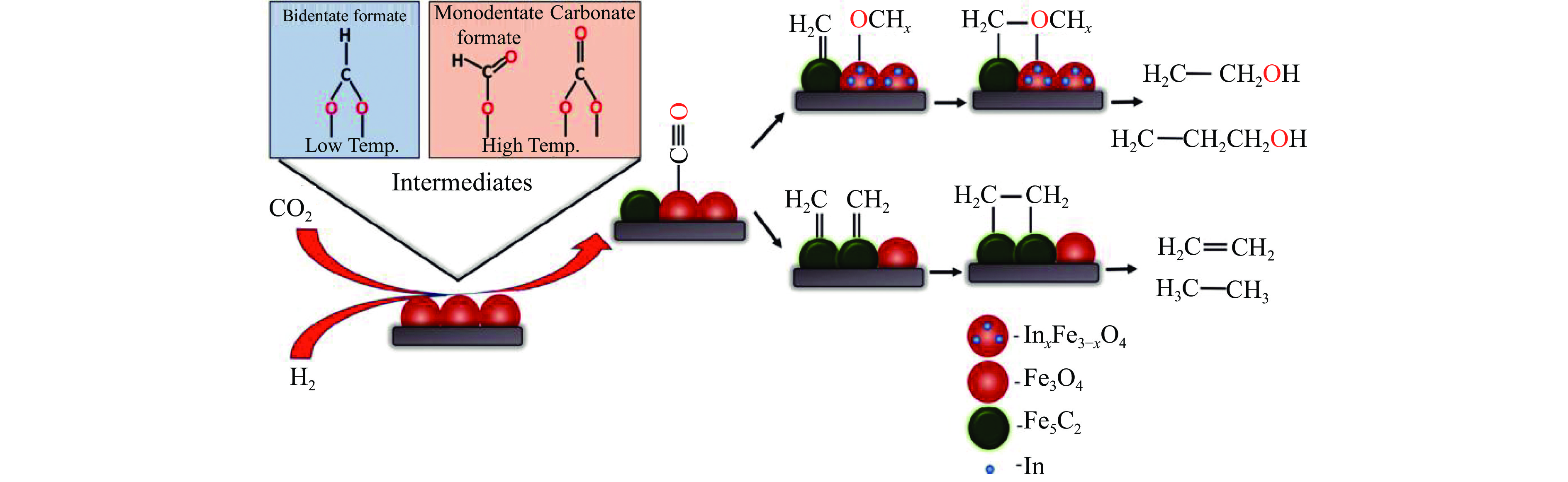
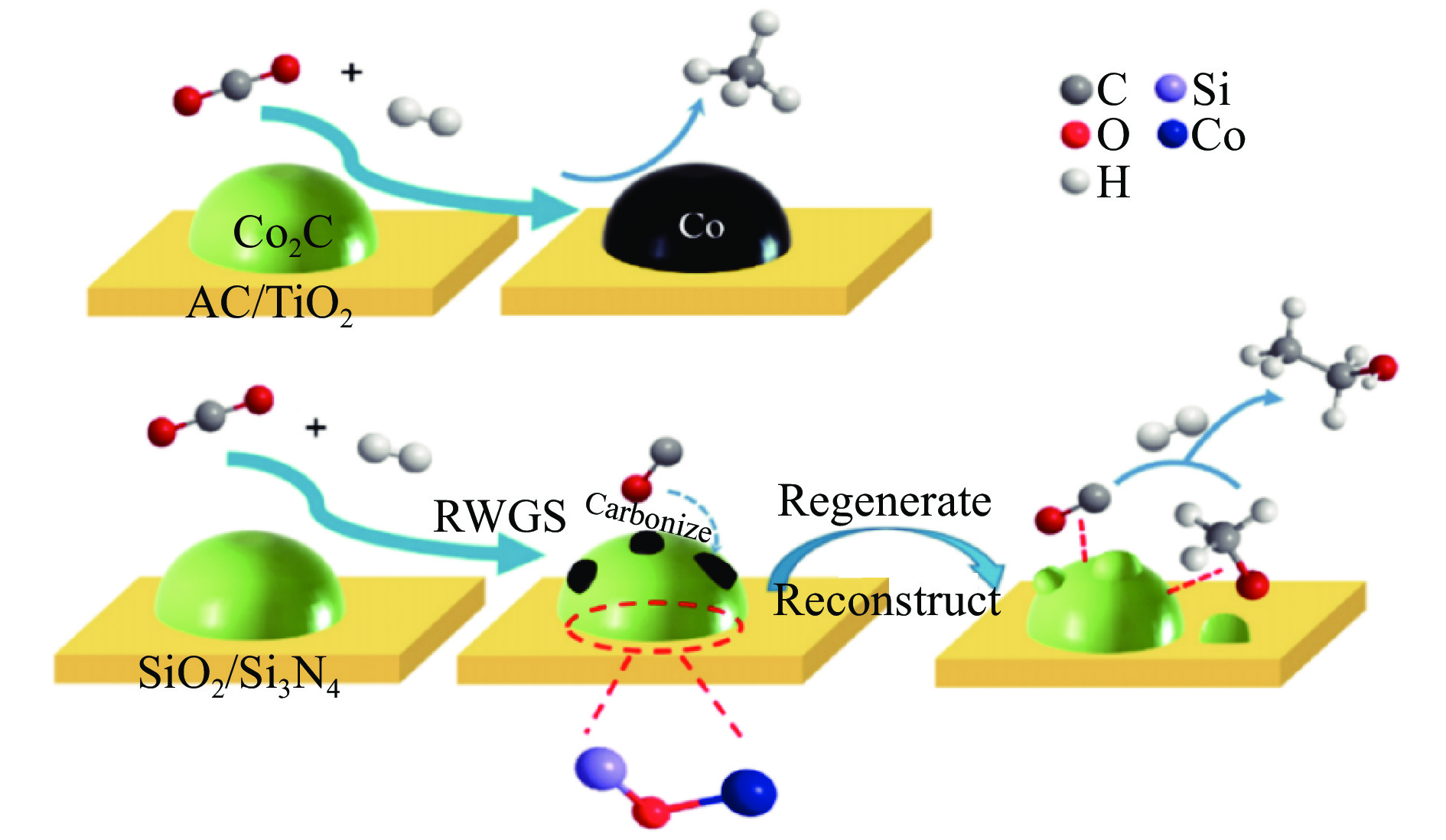

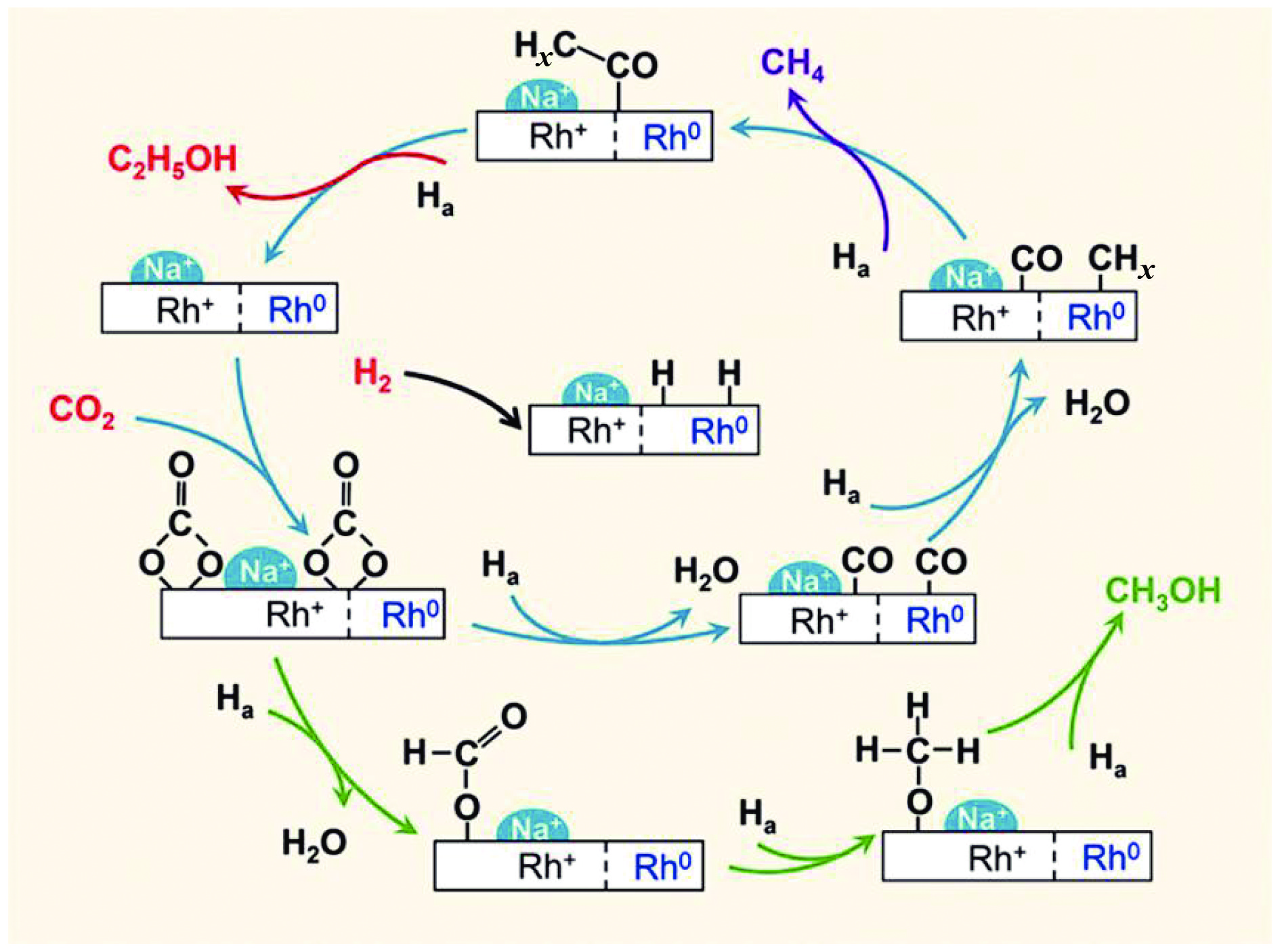


2024, 52(4): 512-524.
doi: 10.1016/S1872-5813(23)60397-4
摘要:
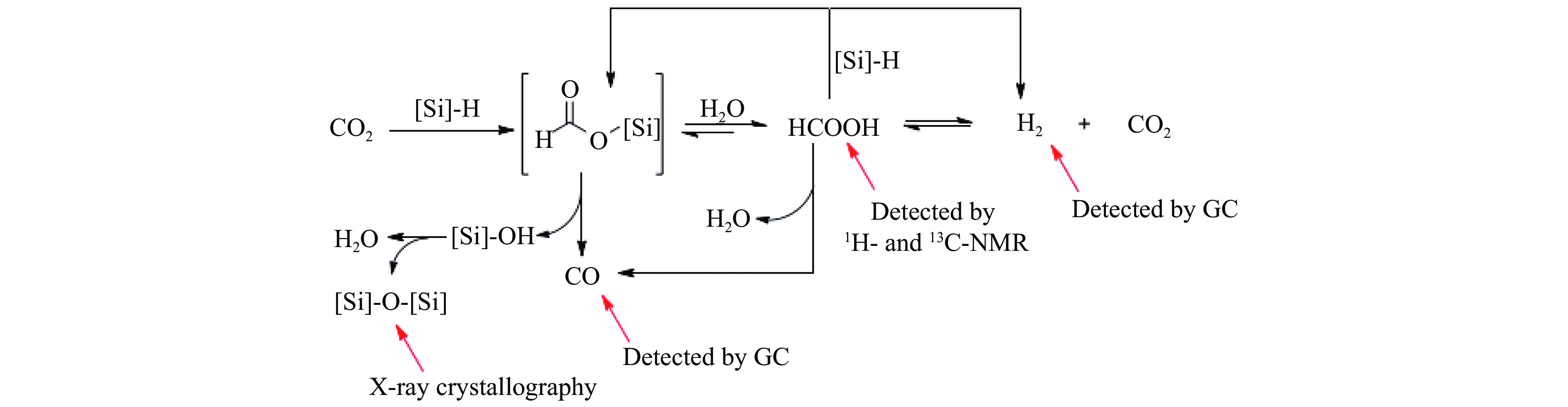
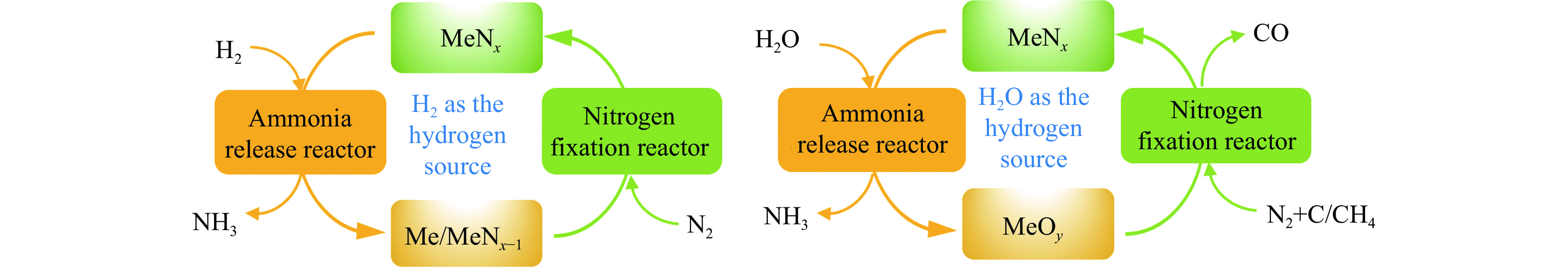
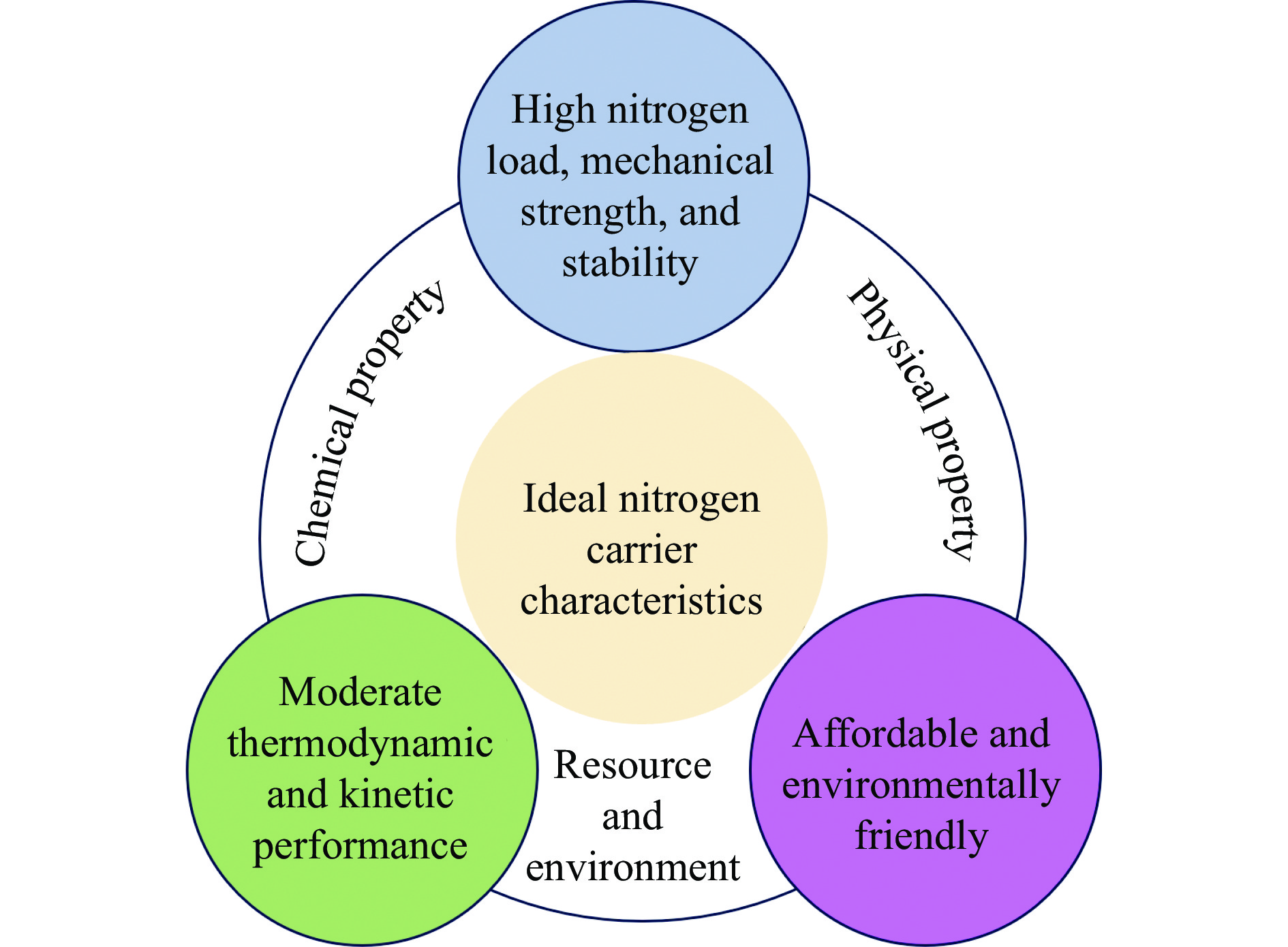
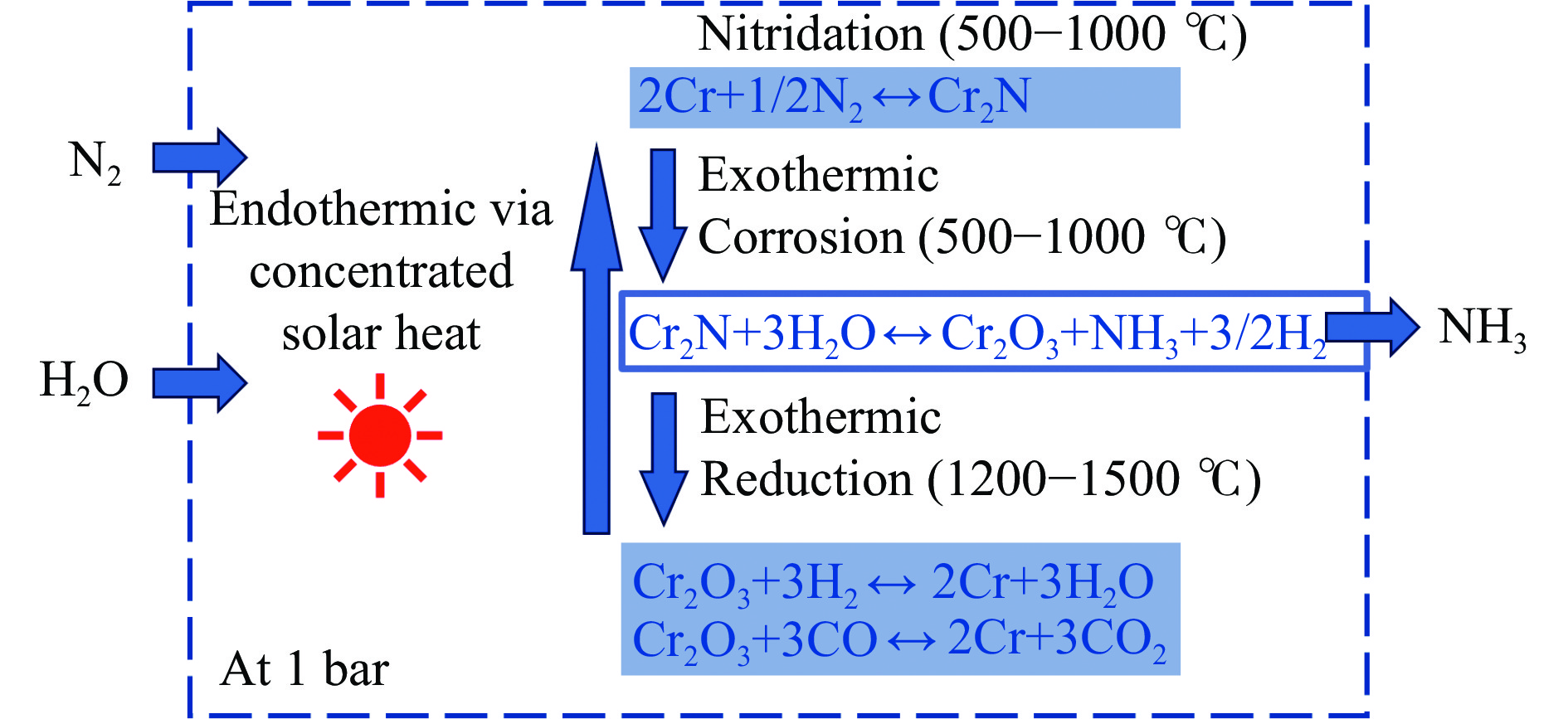
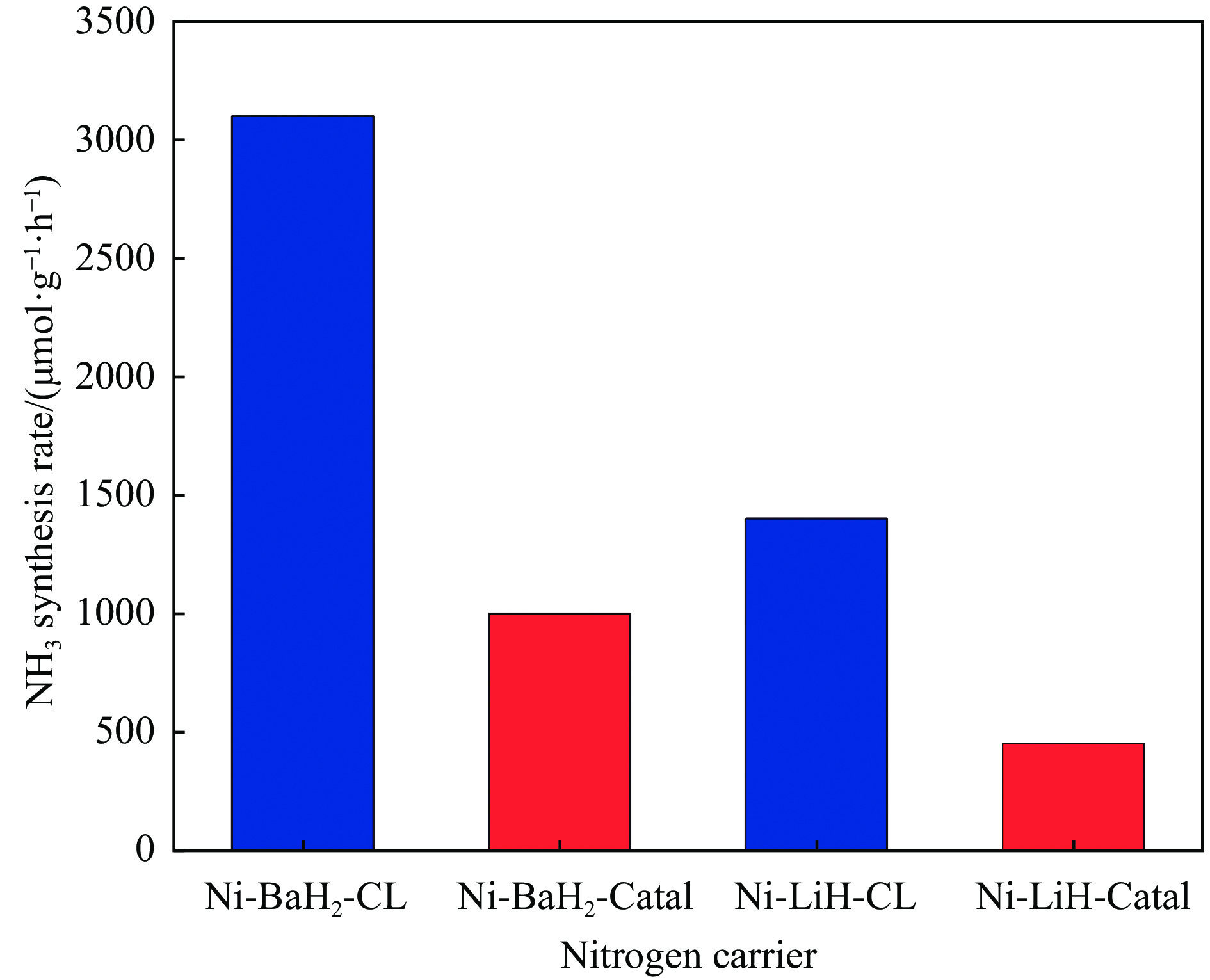
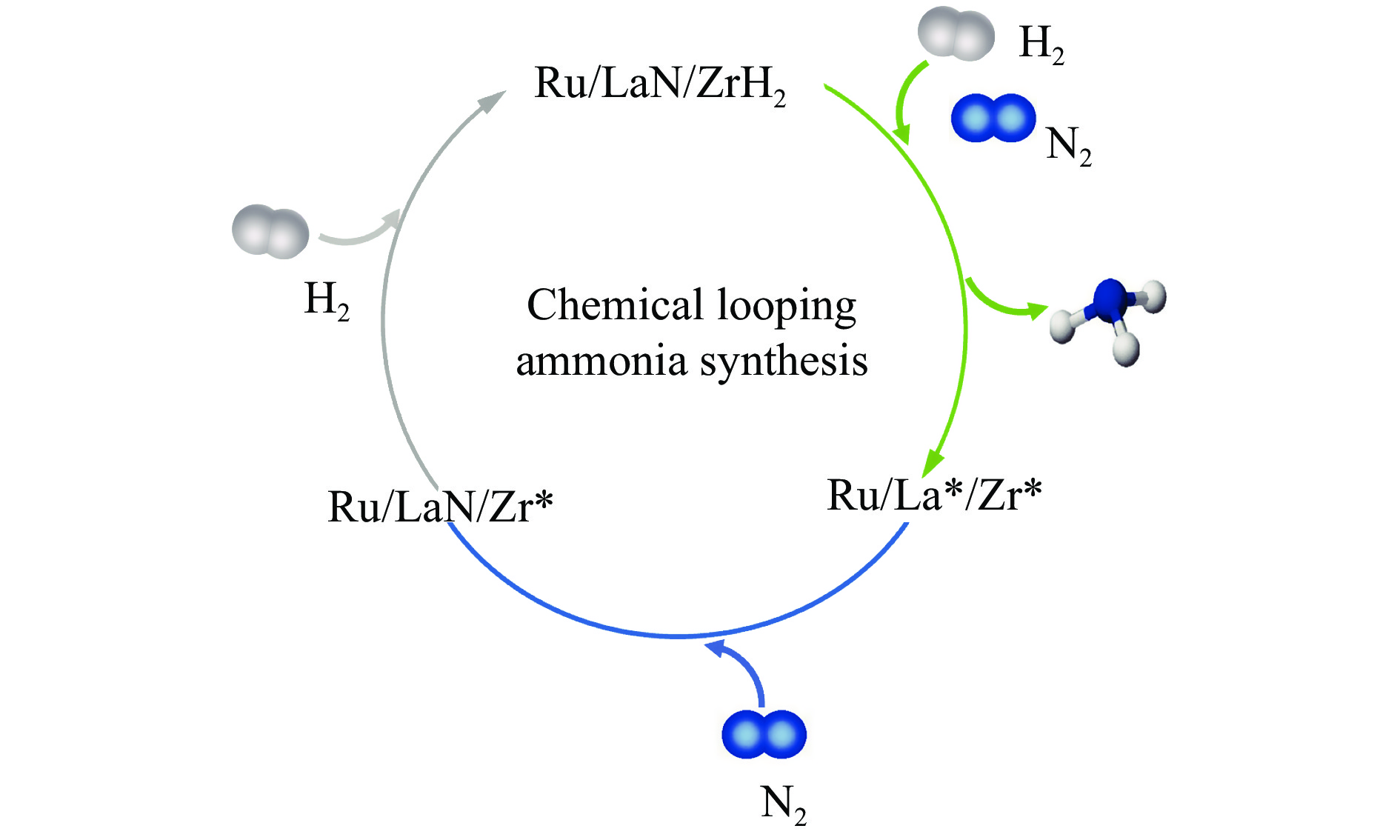
氨不仅是氮肥生产的主要原料,也是可再生能源储存与转化过程中的能源载体之一。因此,开发温和条件下的合成氨技术是近年来重要的研究课题。化学链合成氨技术通过载氮体的传递作用,将合成氨反应解耦为固氮与释氨等多步反应,具有操作简便、反应温和、能耗低等优点。载氮体作为化学链合成氨的关键,起到传递能量及氮物种的作用,目前,载氮体固氮效率低,极大地限制了化学链合成氨技术的发展。基于此,本工作对化学链合成氨中载氮体的设计与应用研究进行综述。首先,对载氮体的设计理论进行了归纳与总结;其次,介绍了载氮体的研究现状,重点对如何提高载氮体的产氨速率以及如何提升晶格氮利用率等问题进行了综述;最后,对化学链合成氨技术所面临的机遇与挑战进行了研究,为今后载氮体的设计与开发提供了参考依据。
氨不仅是氮肥生产的主要原料,也是可再生能源储存与转化过程中的能源载体之一。因此,开发温和条件下的合成氨技术是近年来重要的研究课题。化学链合成氨技术通过载氮体的传递作用,将合成氨反应解耦为固氮与释氨等多步反应,具有操作简便、反应温和、能耗低等优点。载氮体作为化学链合成氨的关键,起到传递能量及氮物种的作用,目前,载氮体固氮效率低,极大地限制了化学链合成氨技术的发展。基于此,本工作对化学链合成氨中载氮体的设计与应用研究进行综述。首先,对载氮体的设计理论进行了归纳与总结;其次,介绍了载氮体的研究现状,重点对如何提高载氮体的产氨速率以及如何提升晶格氮利用率等问题进行了综述;最后,对化学链合成氨技术所面临的机遇与挑战进行了研究,为今后载氮体的设计与开发提供了参考依据。
2024, 52(4): 525-535.
doi: 10.1016/S1872-5813(23)60393-7
摘要:
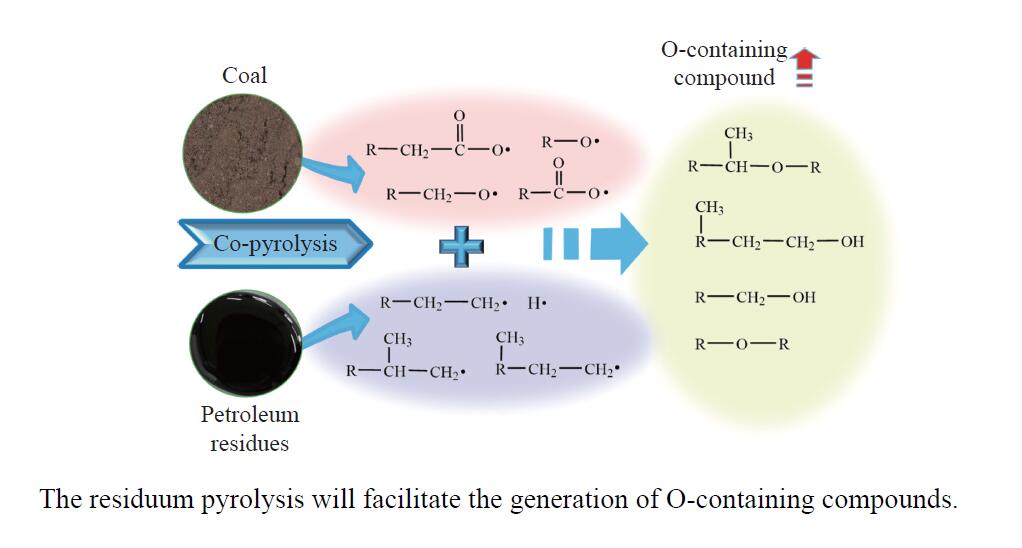
煤油共液化过程中煤与重油先发生共热解,而后加氢转化为小分子产品。因此,阐明重油对煤热解逸出产物的影响规律是调控共液化产物组成的重要热化学基础。本研究采用TG-FTIR对比研究塔河渣油(AR)和淖毛湖煤(NMH)单独热解及其共热解过程,结合热解活化能计算,探索共热解过程中塔河渣油(AR)对淖毛湖煤(NMH)热解产物逸出产物的影响。结果表明,单独热解时AR先于NMH发生热解反应。两者1∶1(质量比)混合共热解时,相比于单独热解计算的理论值,最大失重峰温度前移7 ℃,失重率增加约3%,共热解平均活化能降低23.6 kJ/mol,表明AR率先热解会诱发NMH热解,降低热解反应能垒。TG-FTIR结果显示,AR产生的烷烃类自由基会与NMH热解产生的含氧自由基结合,形成醇、醚等烷基类含氧有机化合物,从而抑制煤中羧基转化为CO2的过程。研究结果有助于揭示共液化反应过程中重油对煤液化产物组成的影响。
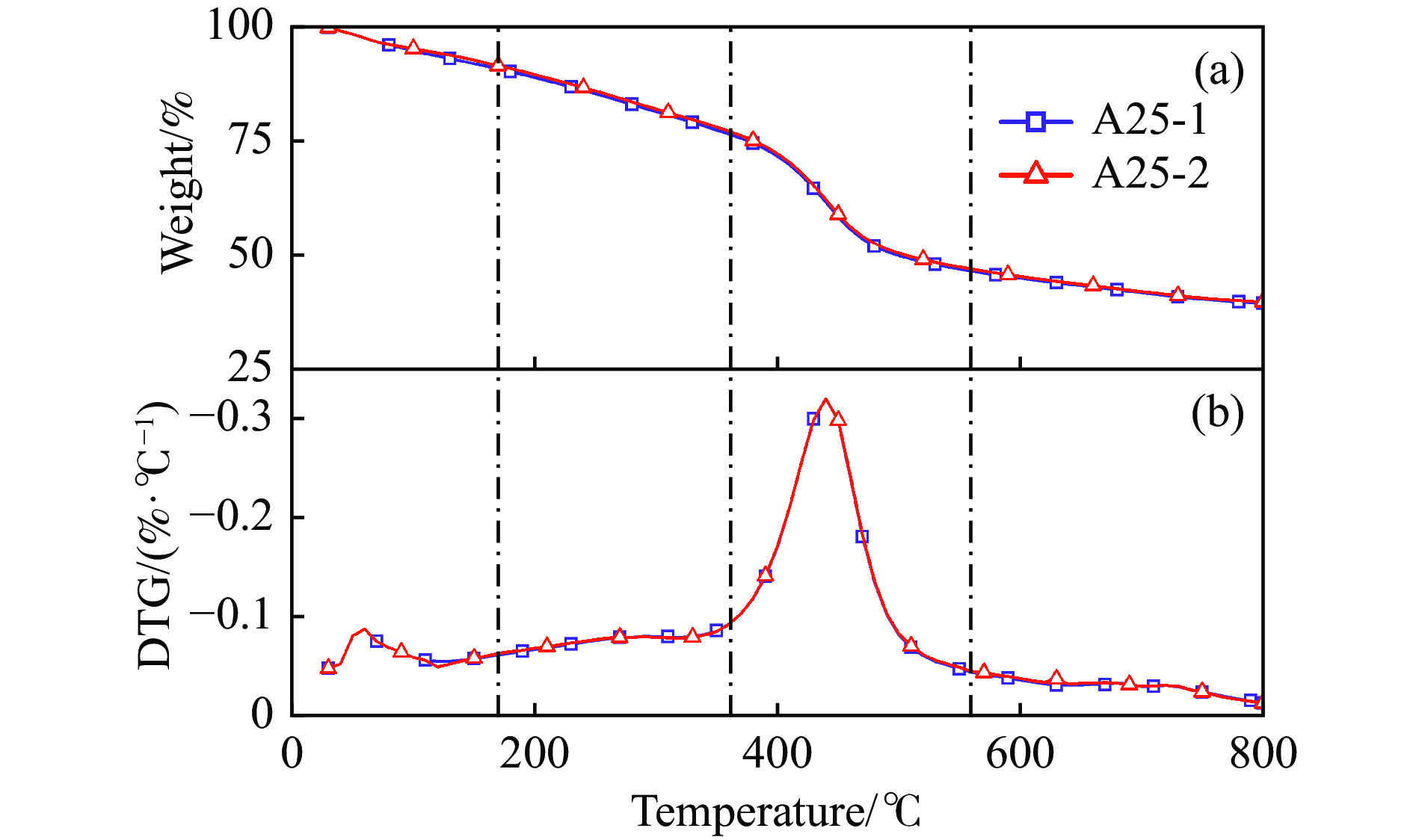
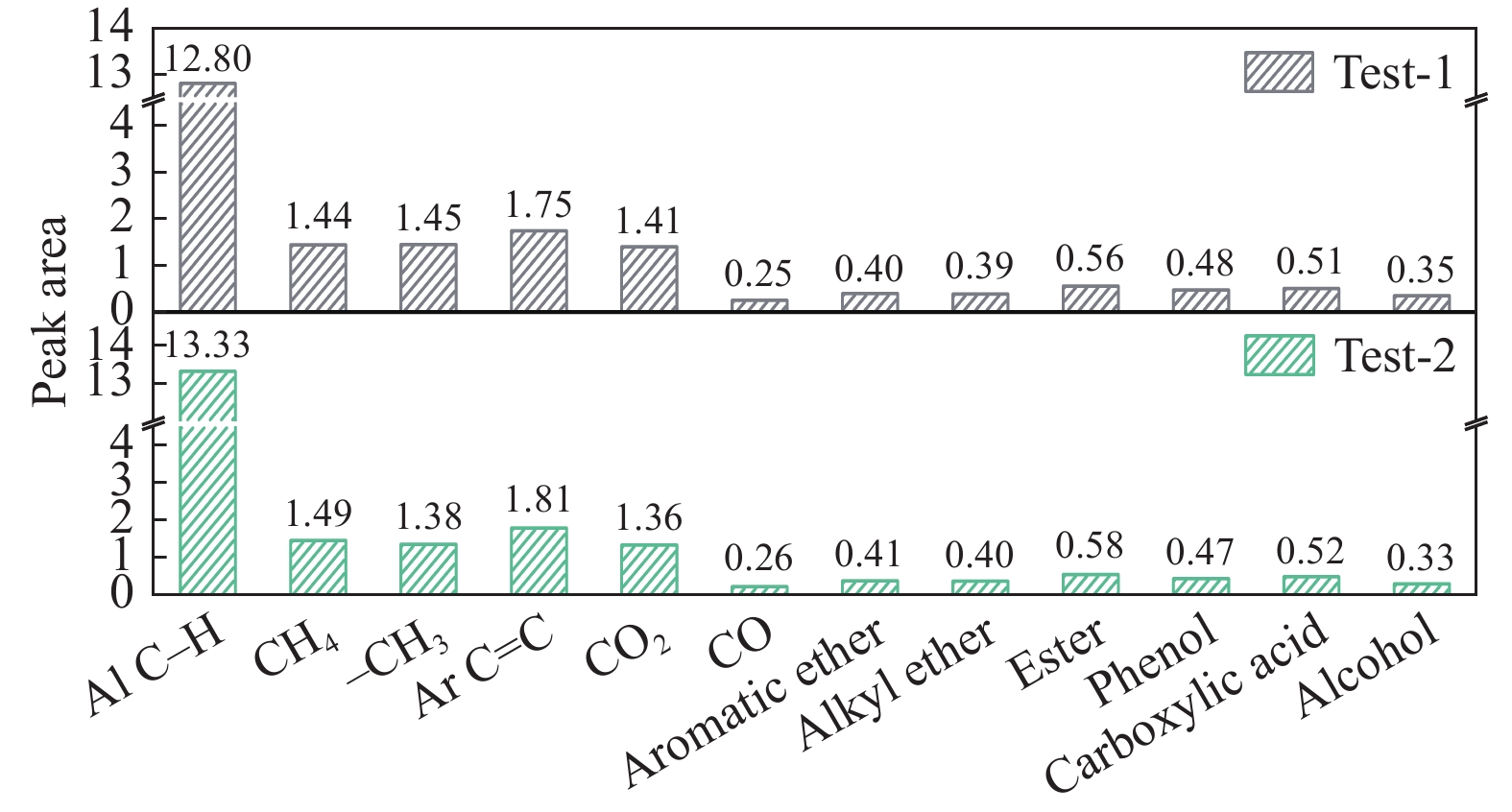
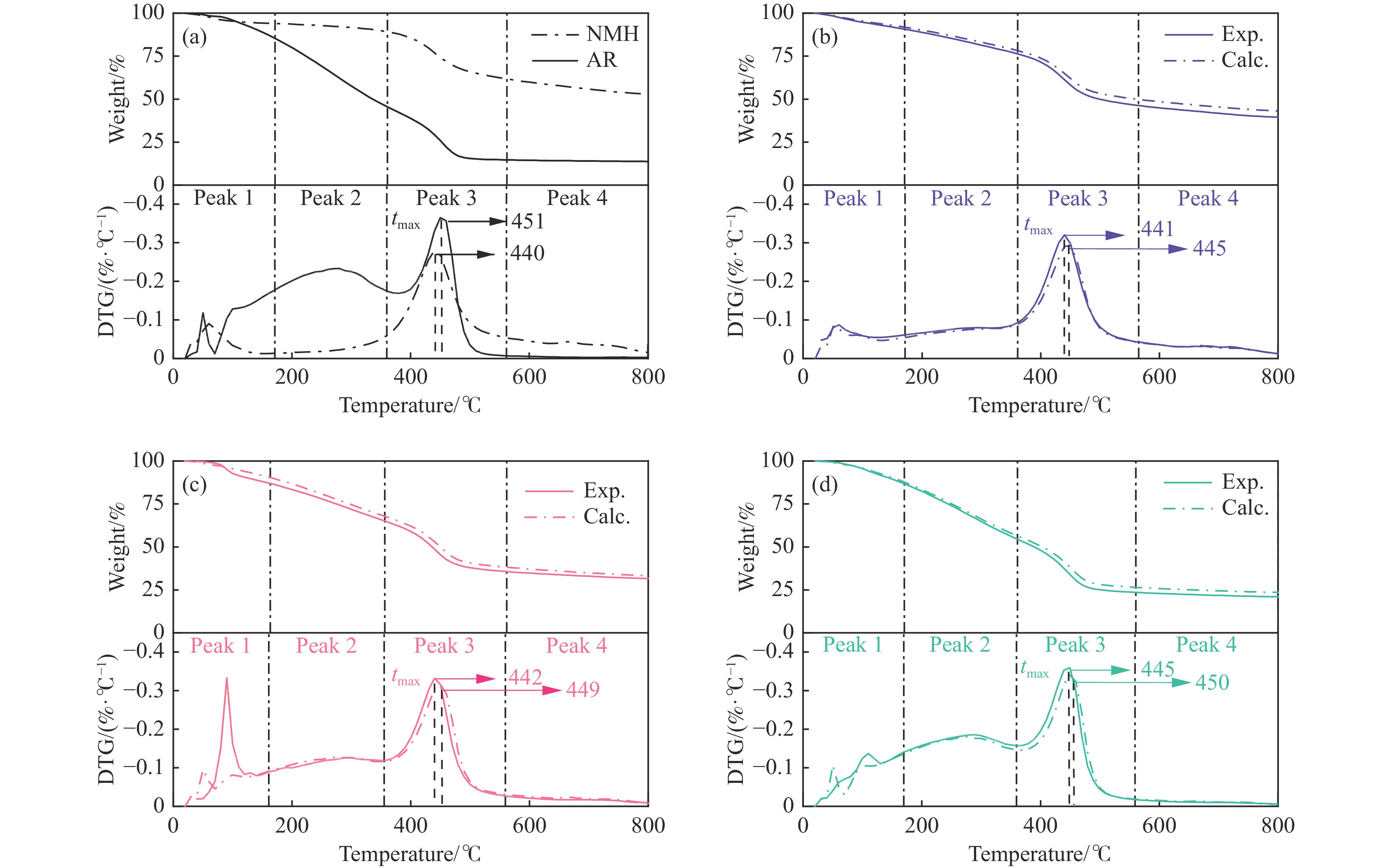
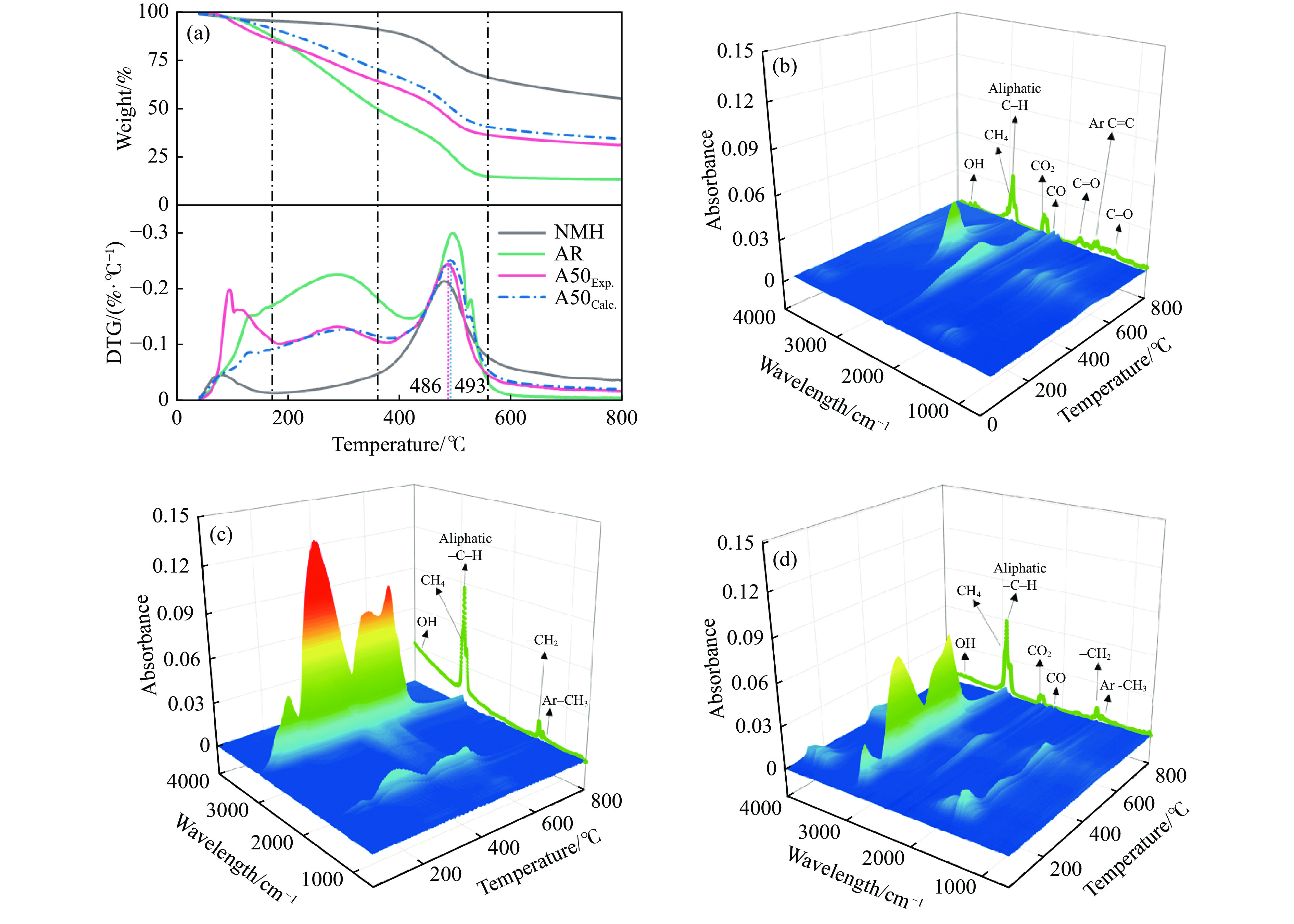
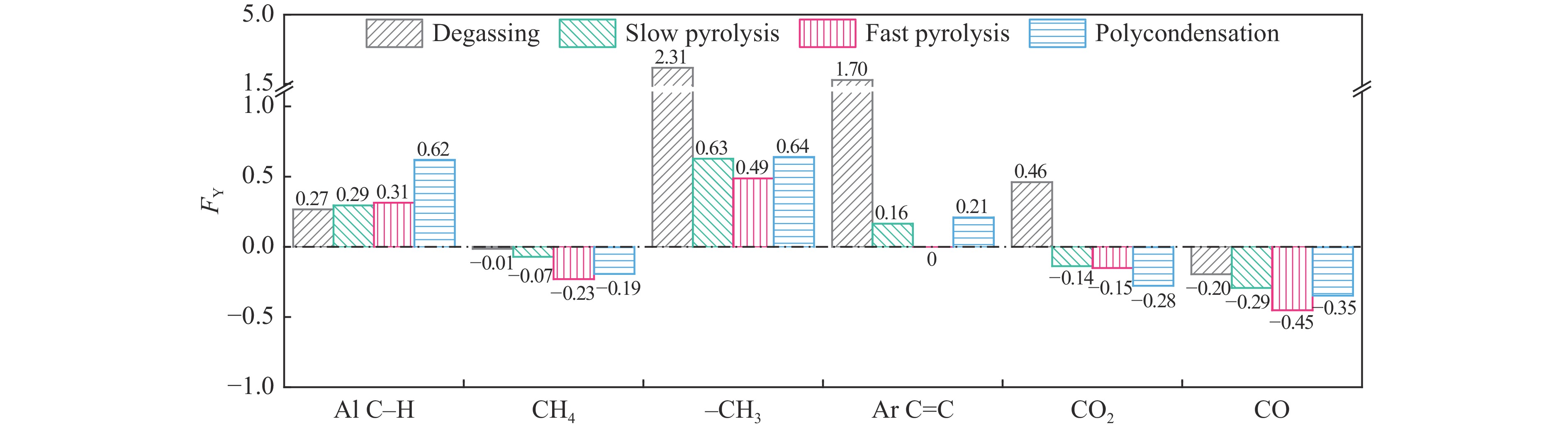
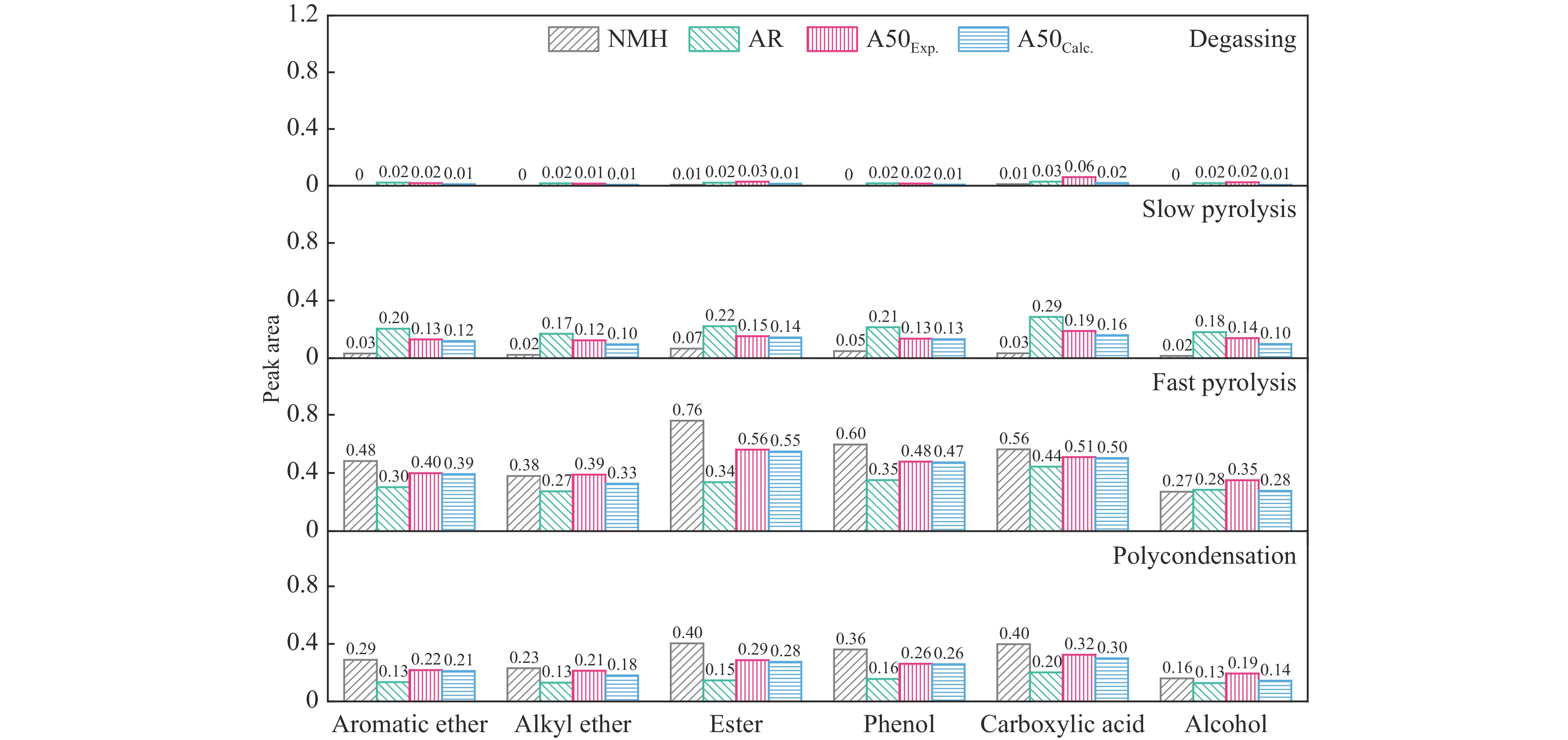
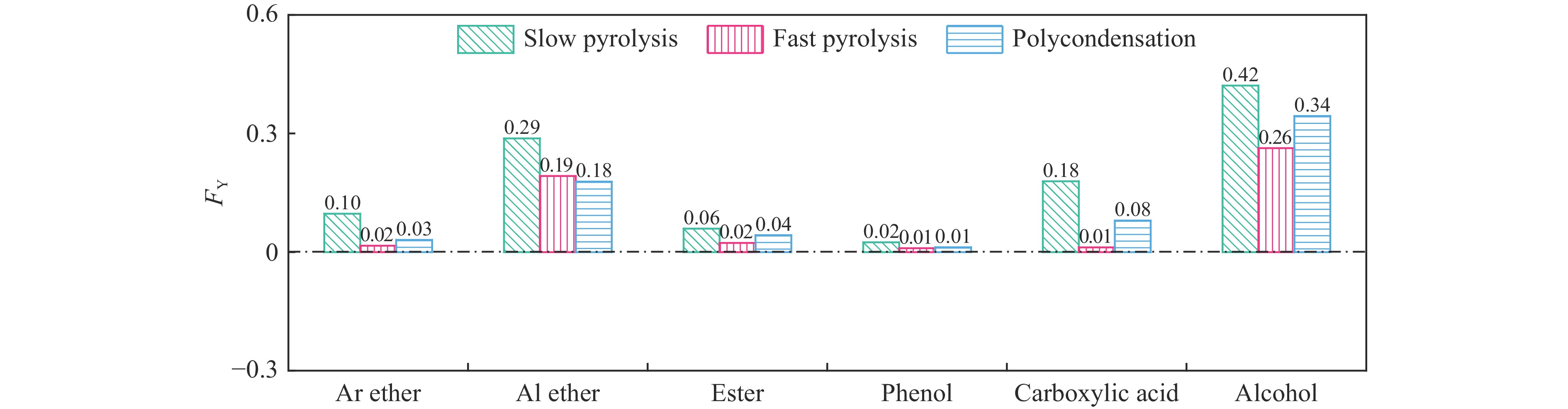
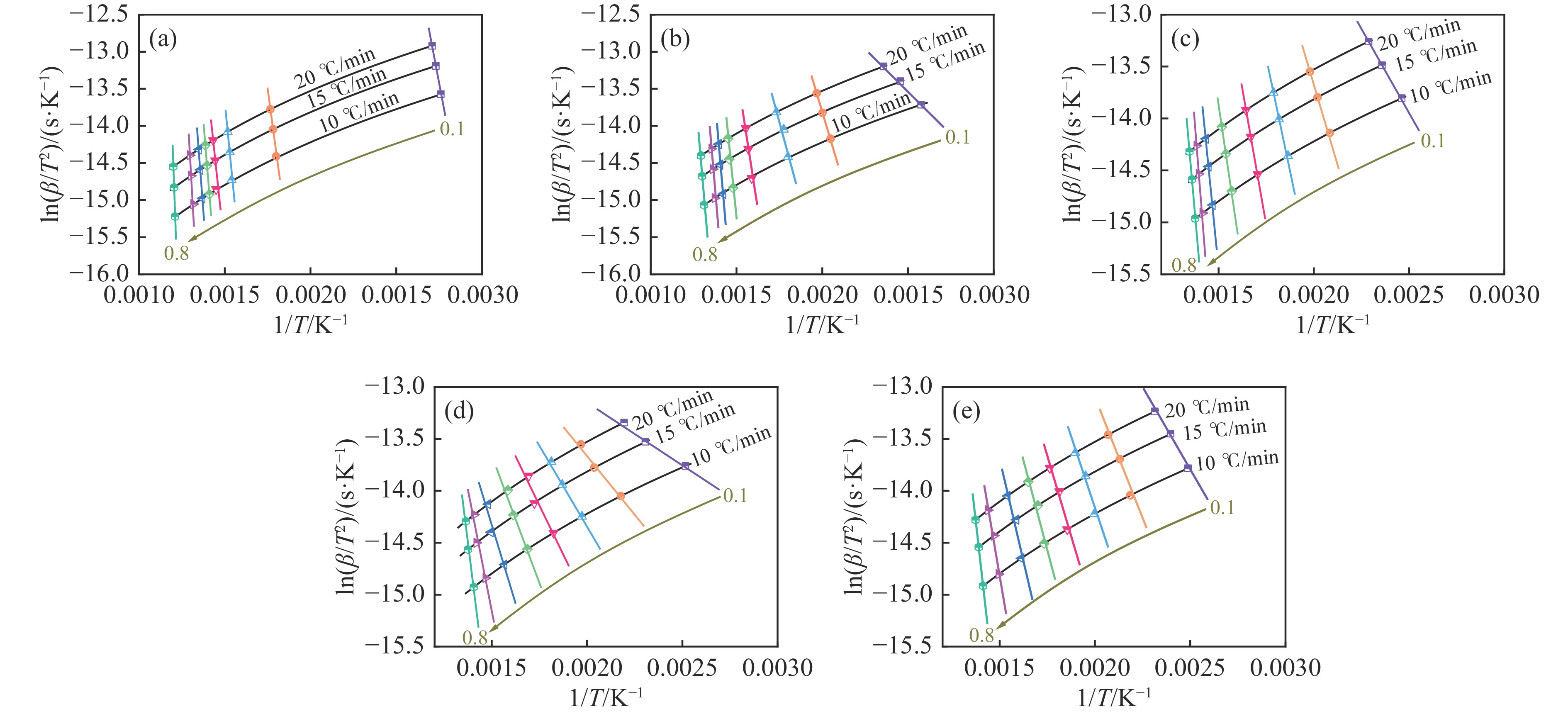
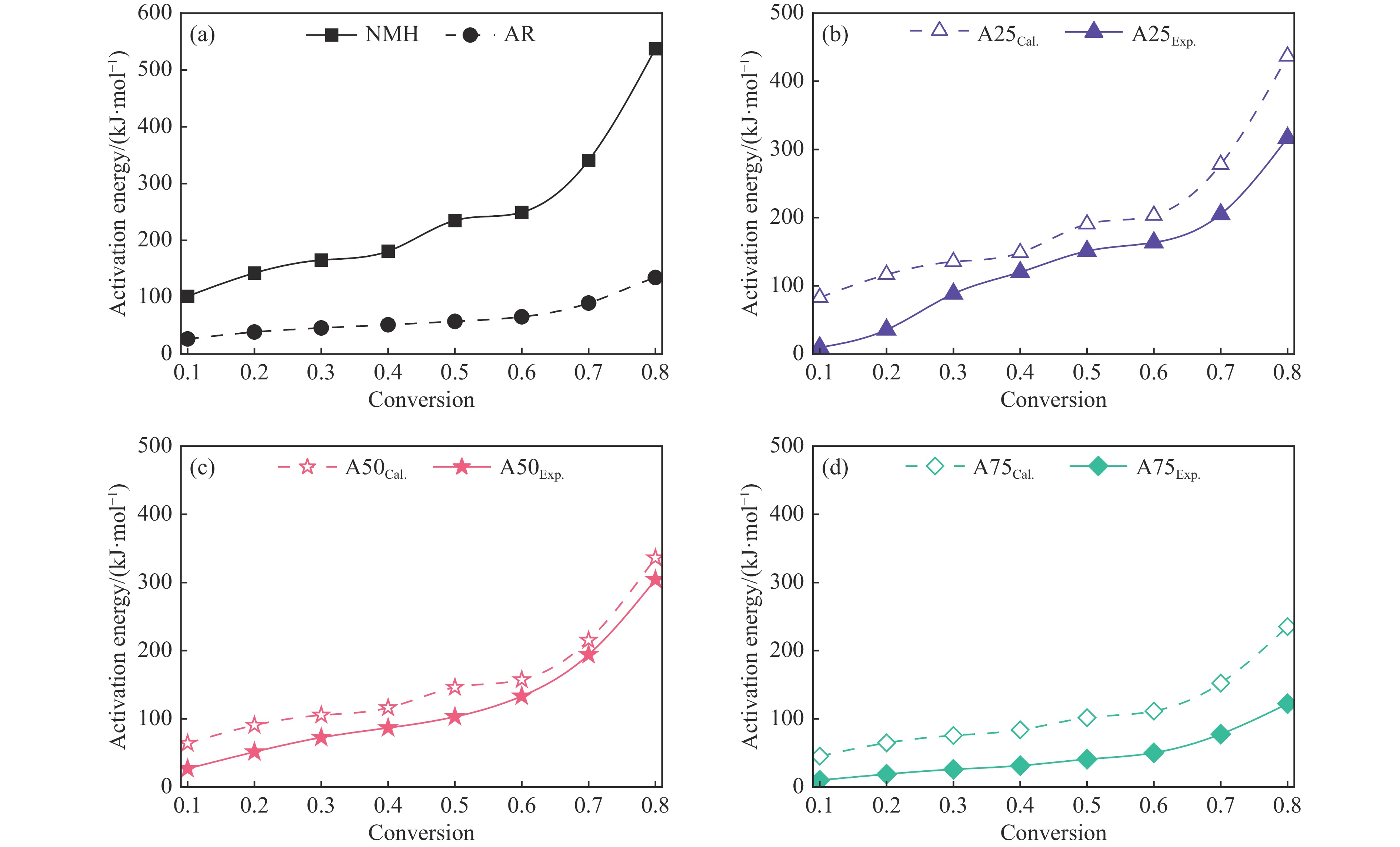
煤油共液化过程中煤与重油先发生共热解,而后加氢转化为小分子产品。因此,阐明重油对煤热解逸出产物的影响规律是调控共液化产物组成的重要热化学基础。本研究采用TG-FTIR对比研究塔河渣油(AR)和淖毛湖煤(NMH)单独热解及其共热解过程,结合热解活化能计算,探索共热解过程中塔河渣油(AR)对淖毛湖煤(NMH)热解产物逸出产物的影响。结果表明,单独热解时AR先于NMH发生热解反应。两者1∶1(质量比)混合共热解时,相比于单独热解计算的理论值,最大失重峰温度前移7 ℃,失重率增加约3%,共热解平均活化能降低23.6 kJ/mol,表明AR率先热解会诱发NMH热解,降低热解反应能垒。TG-FTIR结果显示,AR产生的烷烃类自由基会与NMH热解产生的含氧自由基结合,形成醇、醚等烷基类含氧有机化合物,从而抑制煤中羧基转化为CO2的过程。研究结果有助于揭示共液化反应过程中重油对煤液化产物组成的影响。


2024, 52(4): 536-544.
doi: 10.1016/S1872-5813(23)60387-1
摘要:
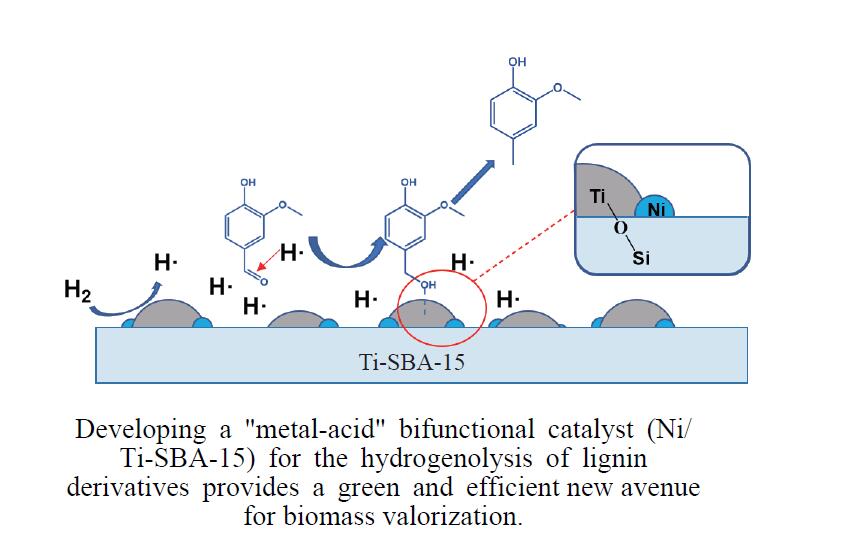
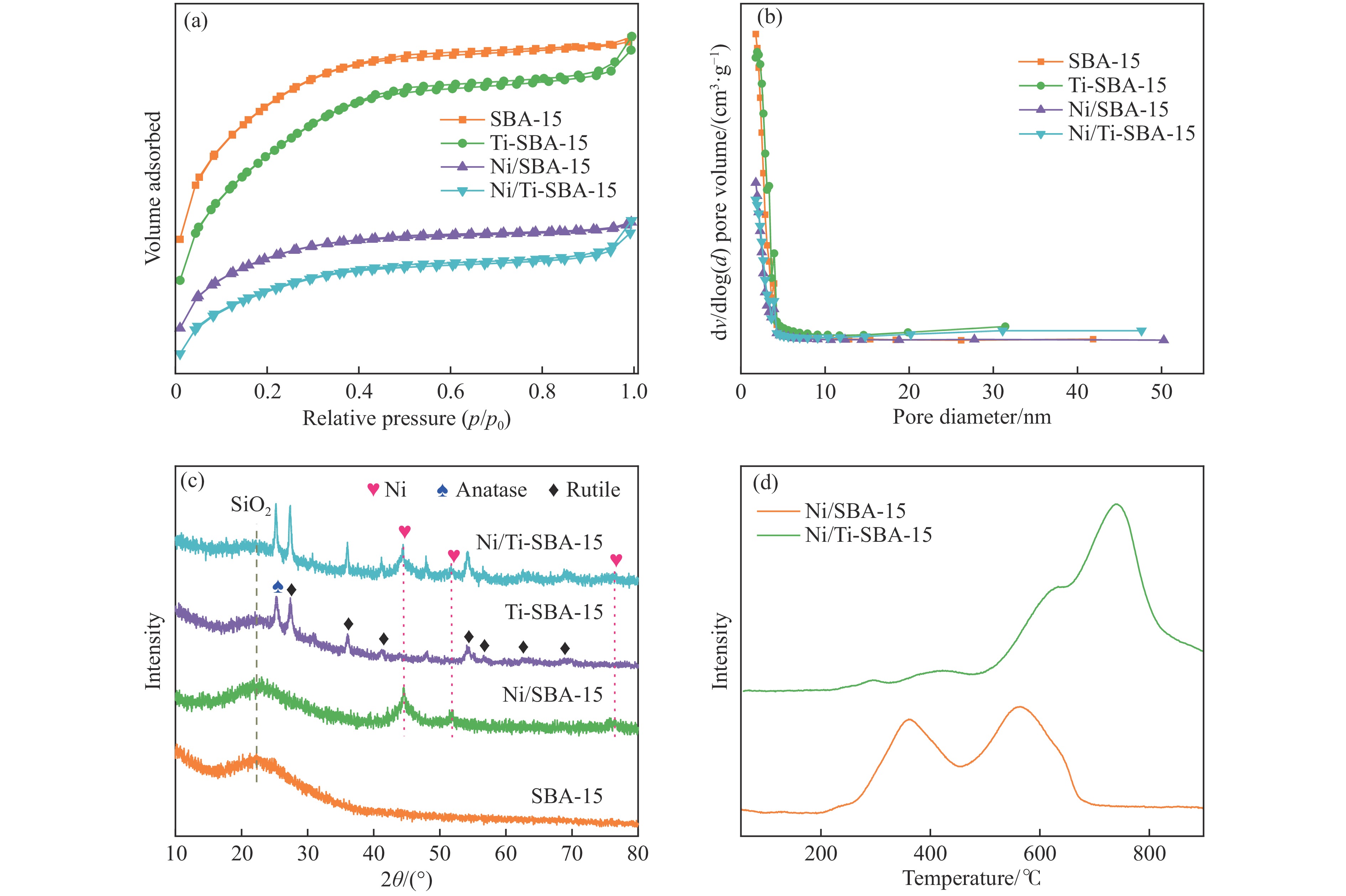
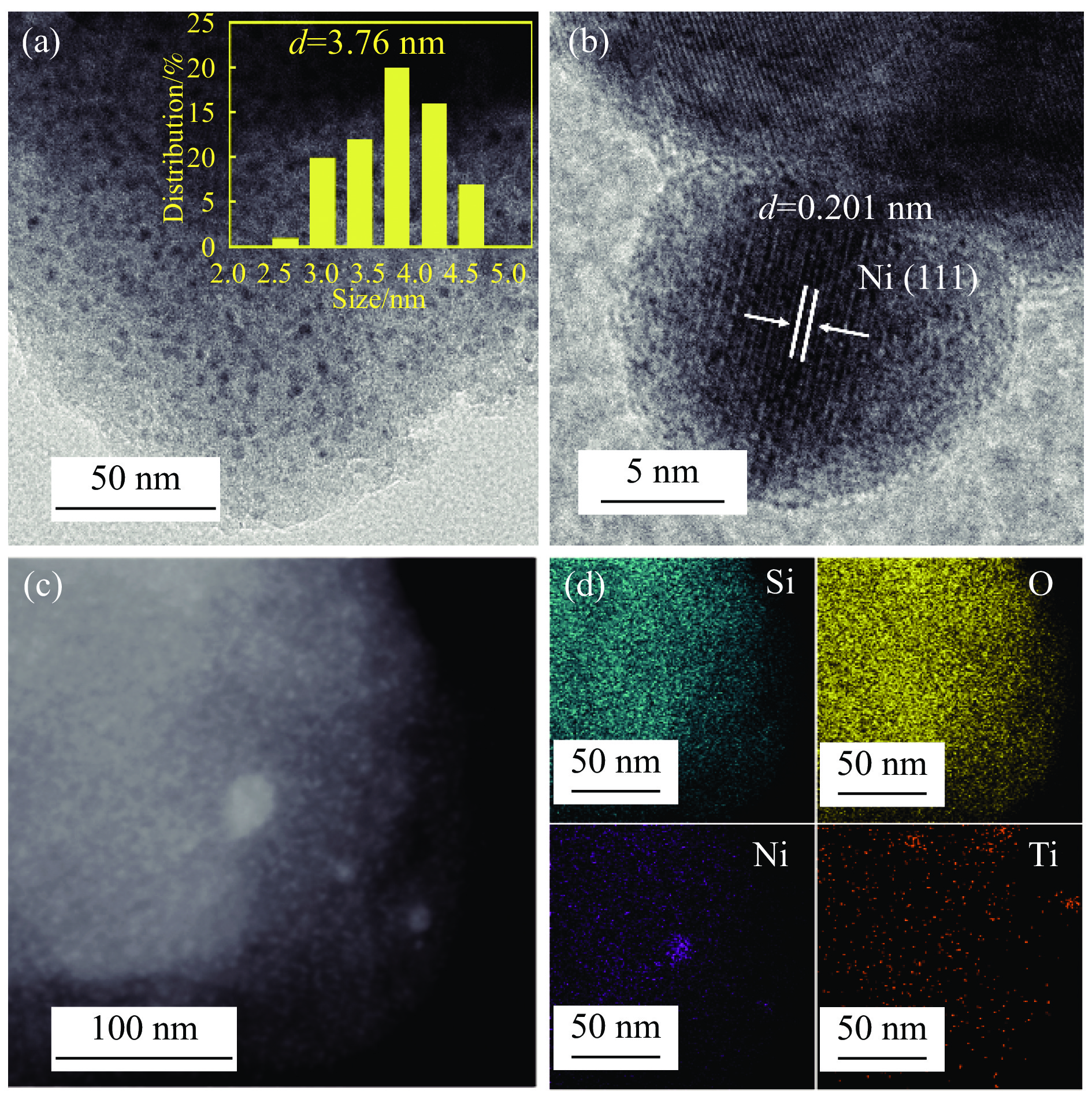
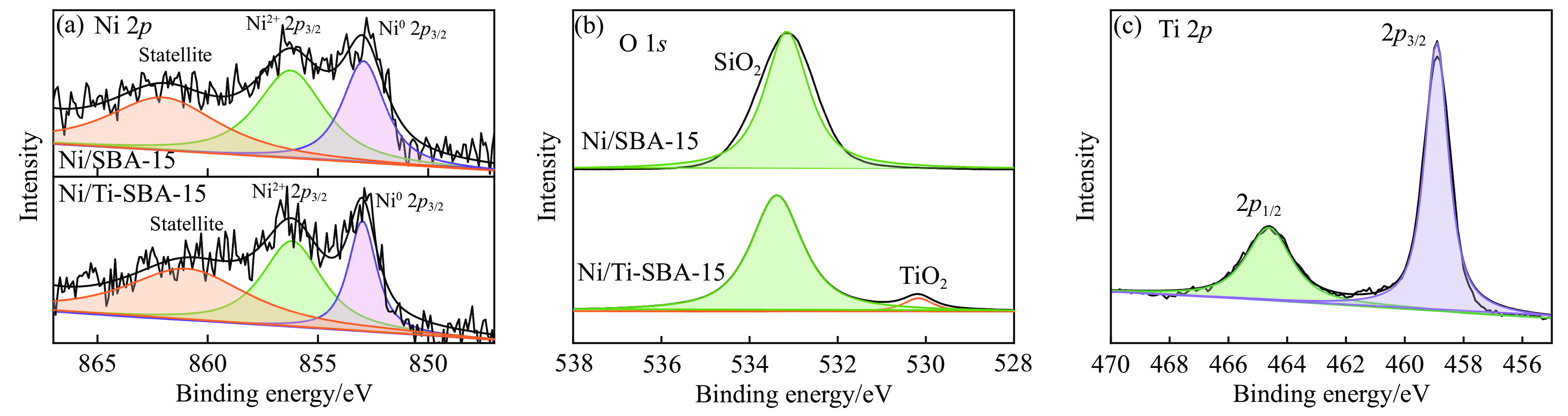
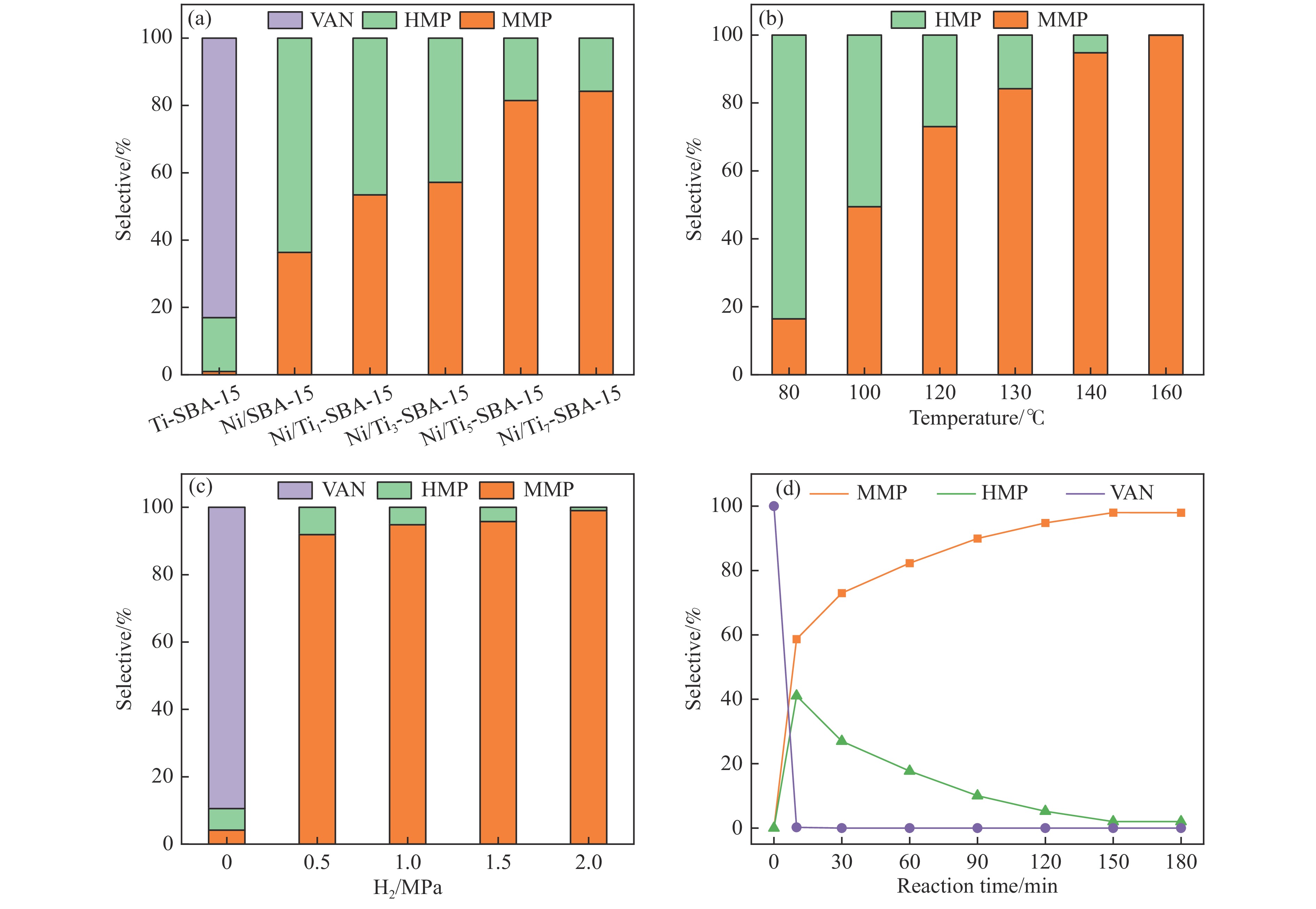
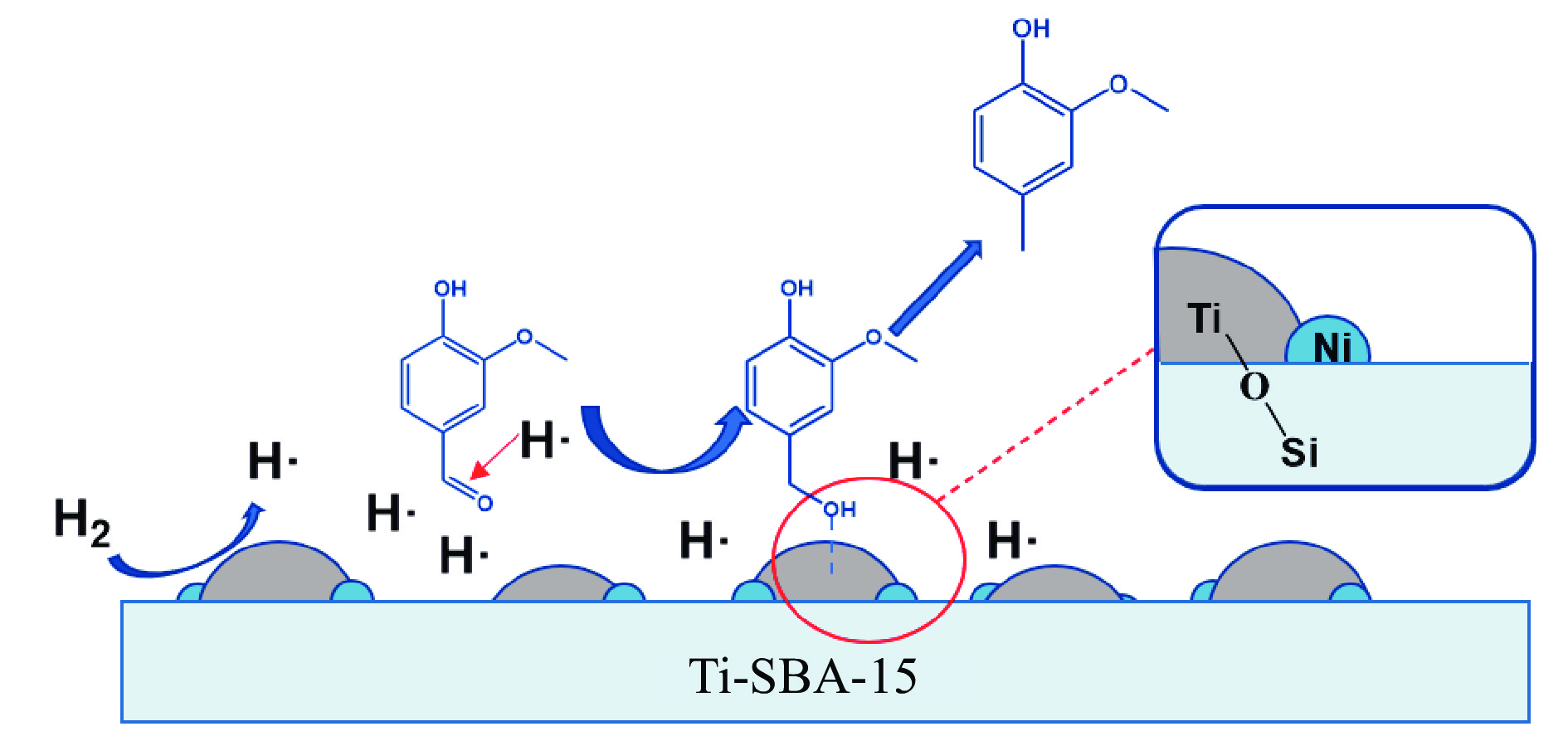
本研究通过在SBA-15分子筛骨架内掺杂Ti物种并负载Ni纳米颗粒合成了“金属-酸”双功能催化剂(Ni/Ti-SBA-15)。Ti的掺杂不仅提高了催化剂酸性位点的数量,还促进了Ni纳米颗粒在载体上的高度分散。在绿色、温和条件下实现了香兰素到2-甲氧基-4-甲基苯酚(MMP)高效转化,目标产物选择性高达96.46%。此外,Ni/Ti-SBA-15催化剂价格低廉,制备工艺简单,这项工作为制备廉价高效催化剂提供了新的思路,有利于实现生物质衍生物的绿色、低成本升级转化。
本研究通过在SBA-15分子筛骨架内掺杂Ti物种并负载Ni纳米颗粒合成了“金属-酸”双功能催化剂(Ni/Ti-SBA-15)。Ti的掺杂不仅提高了催化剂酸性位点的数量,还促进了Ni纳米颗粒在载体上的高度分散。在绿色、温和条件下实现了香兰素到2-甲氧基-4-甲基苯酚(MMP)高效转化,目标产物选择性高达96.46%。此外,Ni/Ti-SBA-15催化剂价格低廉,制备工艺简单,这项工作为制备廉价高效催化剂提供了新的思路,有利于实现生物质衍生物的绿色、低成本升级转化。

2024, 52(4): 545-552.
doi: 10.1016/S1872-5813(23)60394-9
摘要:
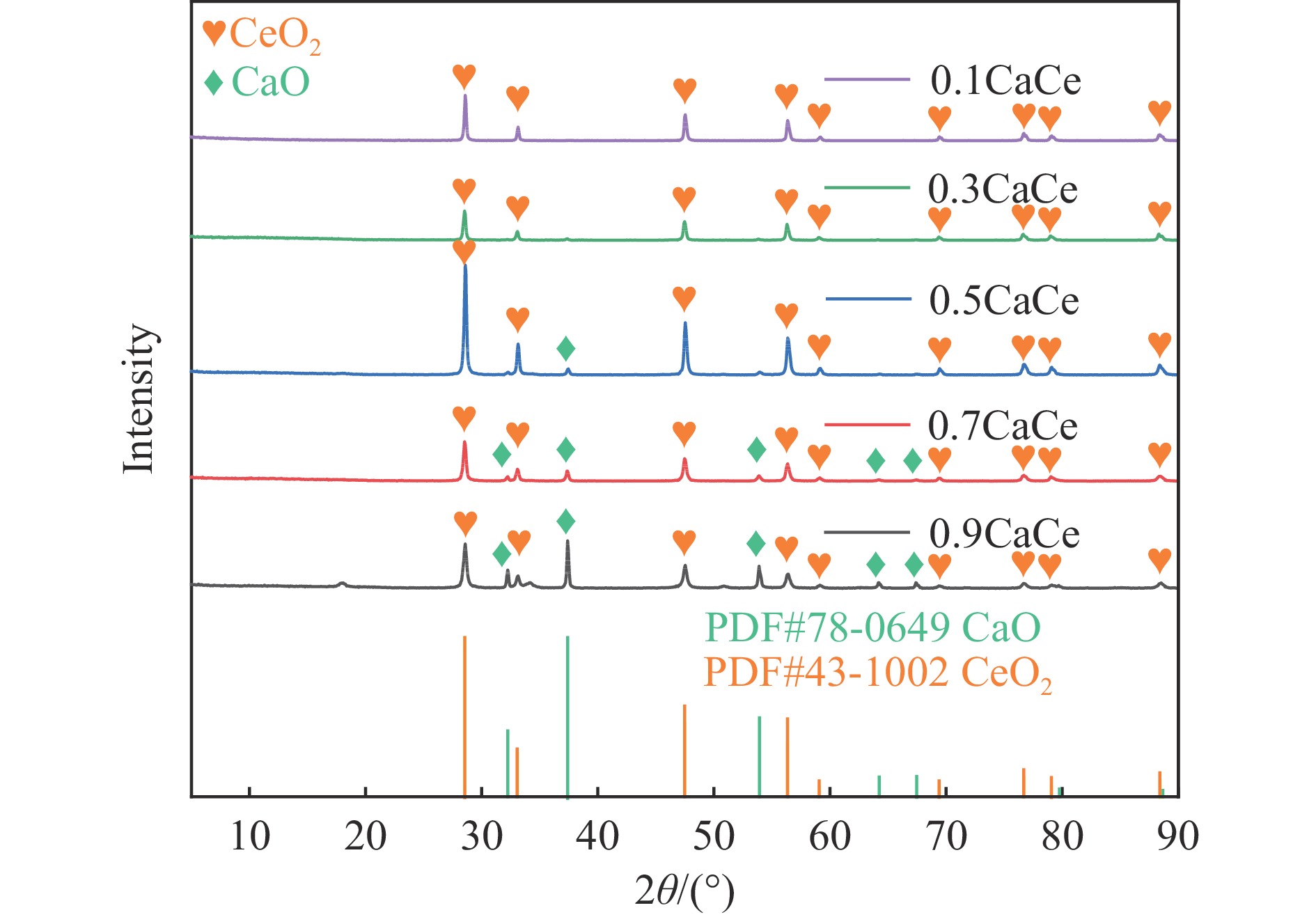
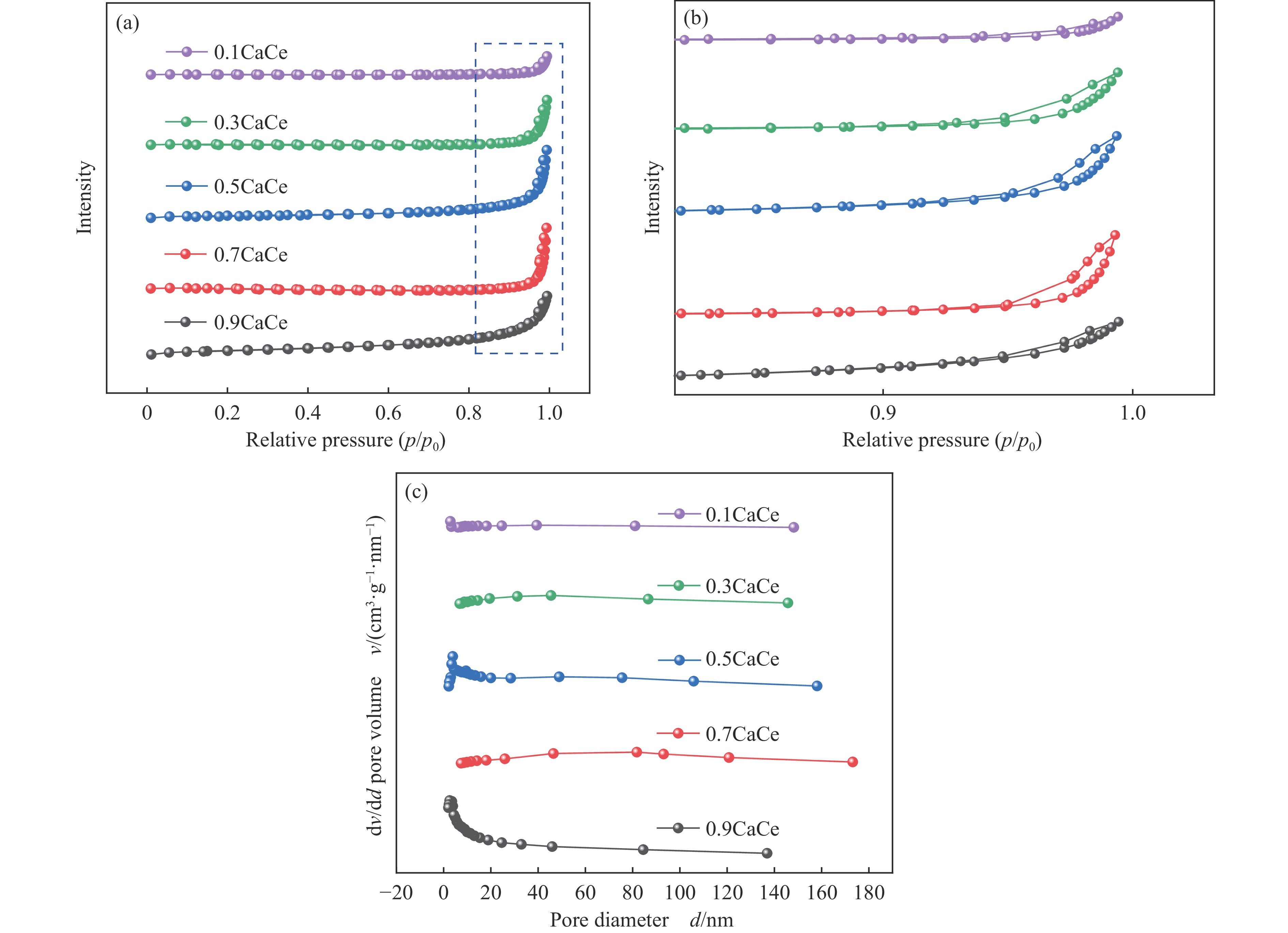
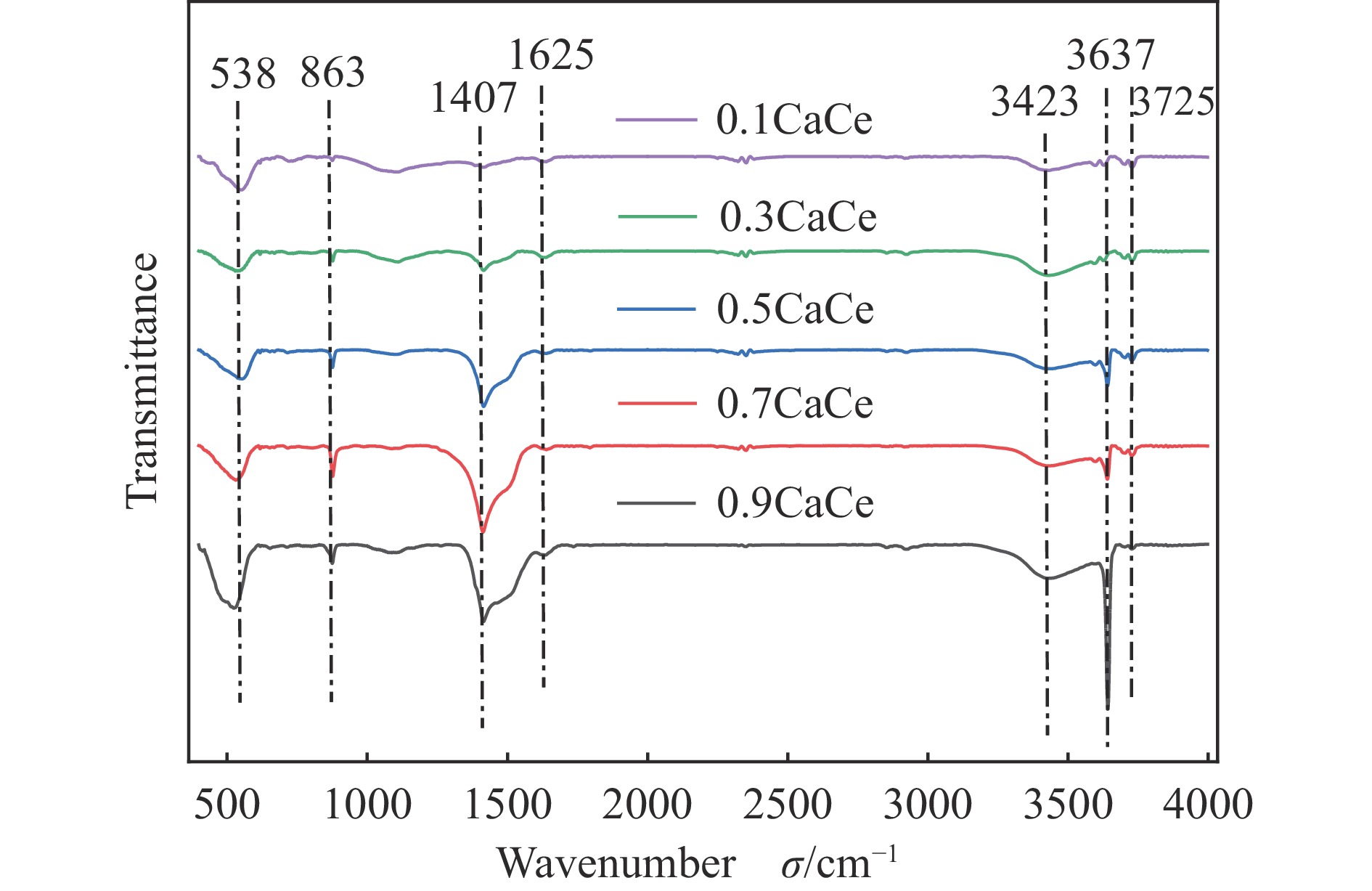
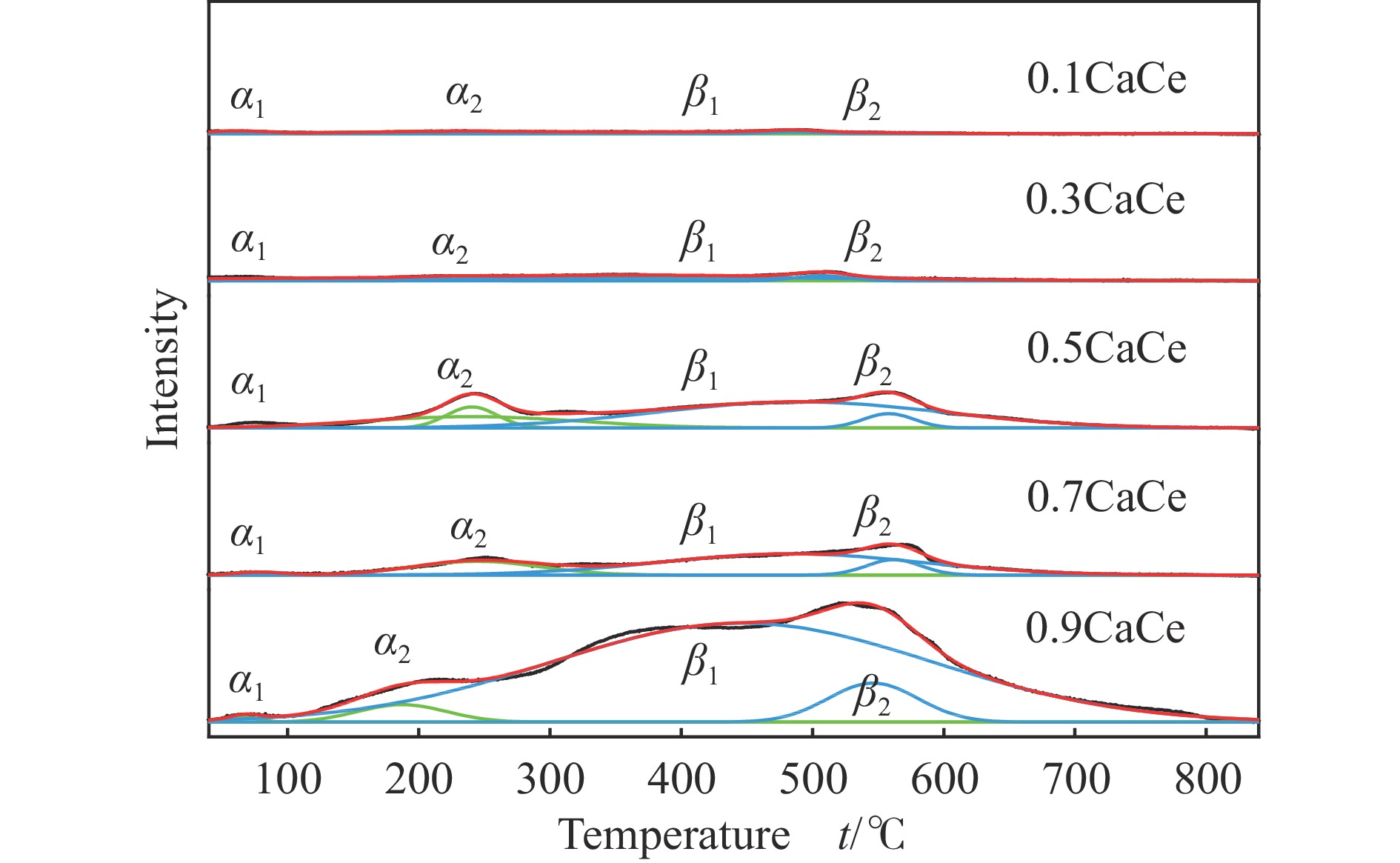
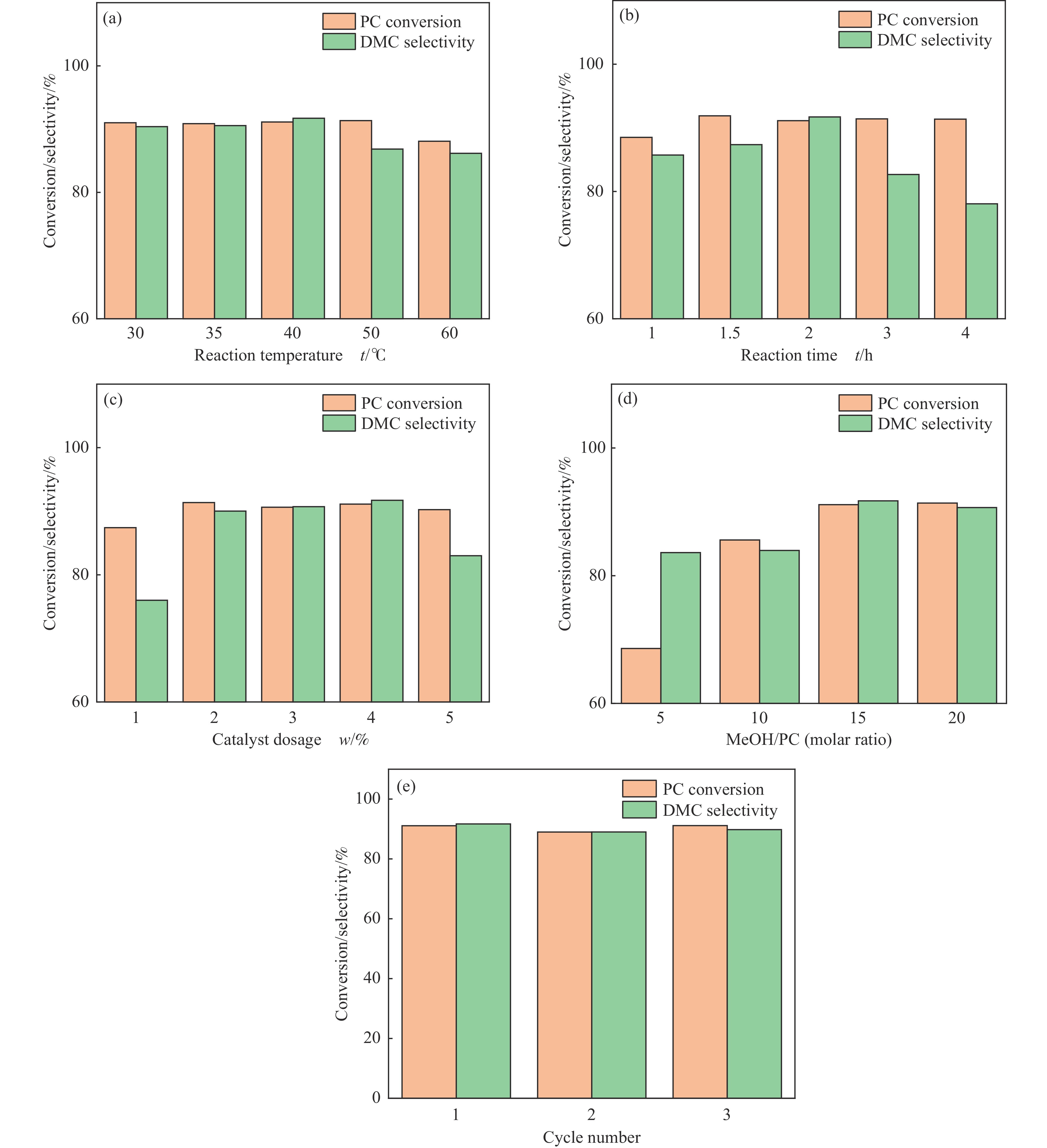
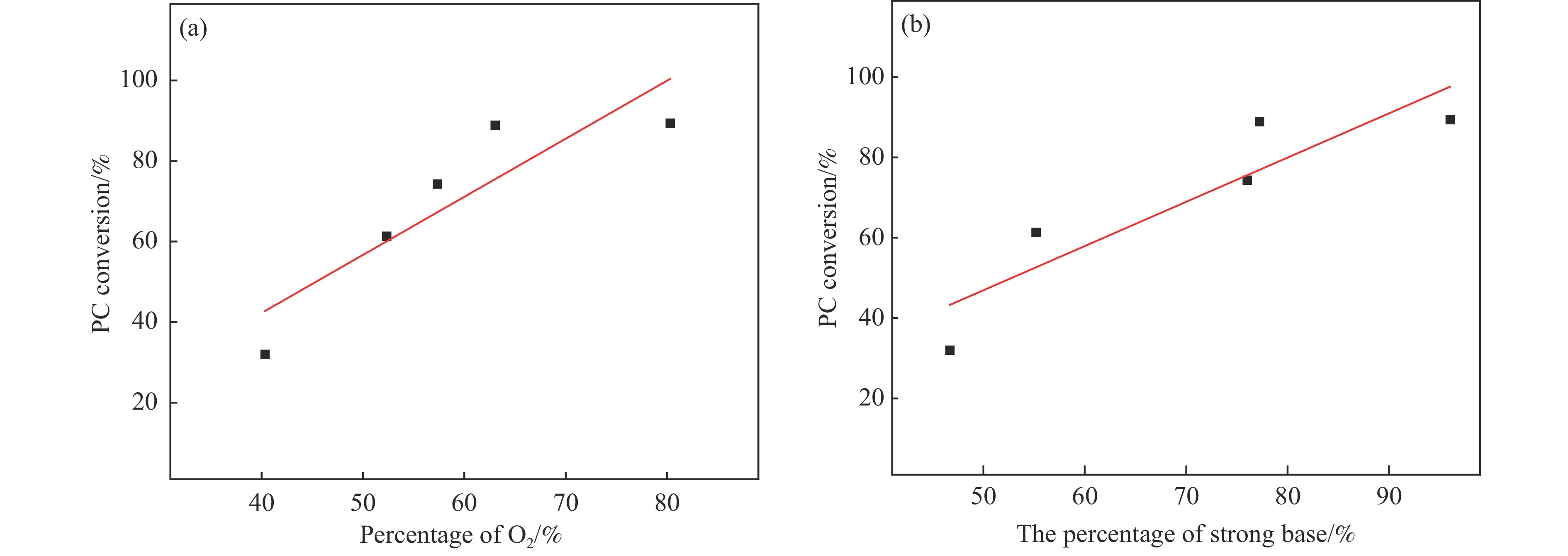
采用溶胶凝胶法制备了不同比例的钙铈基催化剂,并研究了其对于碳酸丙烯酯和甲醇制备碳酸二甲酯的酯交换反应性能。结果表明, Ca∶Ce=9的催化剂在反应时间2 h,温度40 ℃,甲醇与碳酸丙烯酯物质的量比为15∶1,催化剂用量为碳酸丙烯酯用量4%的条件下,碳酸丙烯酯转化率达到91.1%,碳酸二甲酯选择性达到91.7%。采用XRD、N2吸附-脱附、FT-IR、XPS和CO2-TPD对催化剂进行了表征。结果表明,催化剂表面的氧空穴越多,中等碱性位数量越多,越有利于甲醇的活化,催化剂的活性越好。
采用溶胶凝胶法制备了不同比例的钙铈基催化剂,并研究了其对于碳酸丙烯酯和甲醇制备碳酸二甲酯的酯交换反应性能。结果表明, Ca∶Ce=9的催化剂在反应时间2 h,温度40 ℃,甲醇与碳酸丙烯酯物质的量比为15∶1,催化剂用量为碳酸丙烯酯用量4%的条件下,碳酸丙烯酯转化率达到91.1%,碳酸二甲酯选择性达到91.7%。采用XRD、N2吸附-脱附、FT-IR、XPS和CO2-TPD对催化剂进行了表征。结果表明,催化剂表面的氧空穴越多,中等碱性位数量越多,越有利于甲醇的活化,催化剂的活性越好。

2024, 52(4): 553-564.
doi: 10.1016/S1872-5813(23)60399-8
摘要:
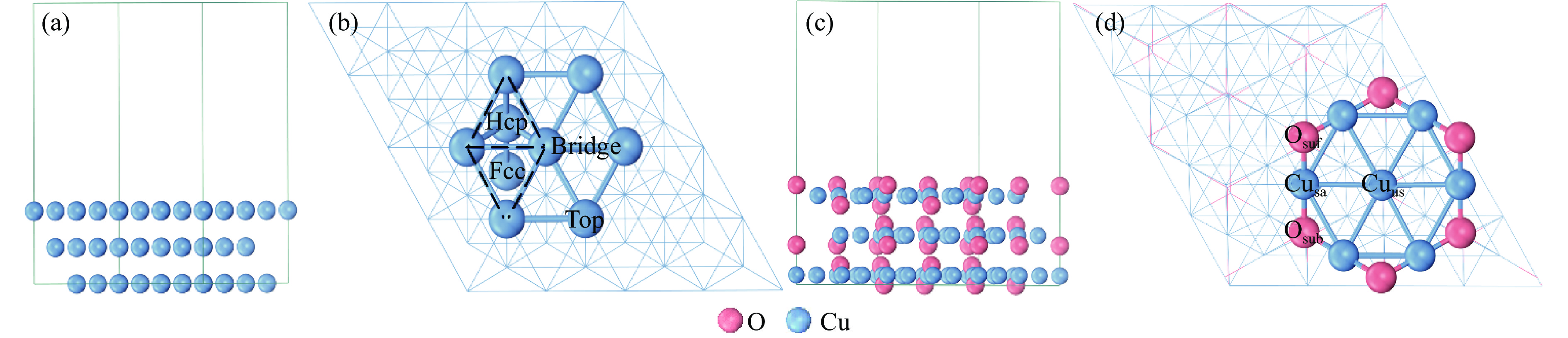
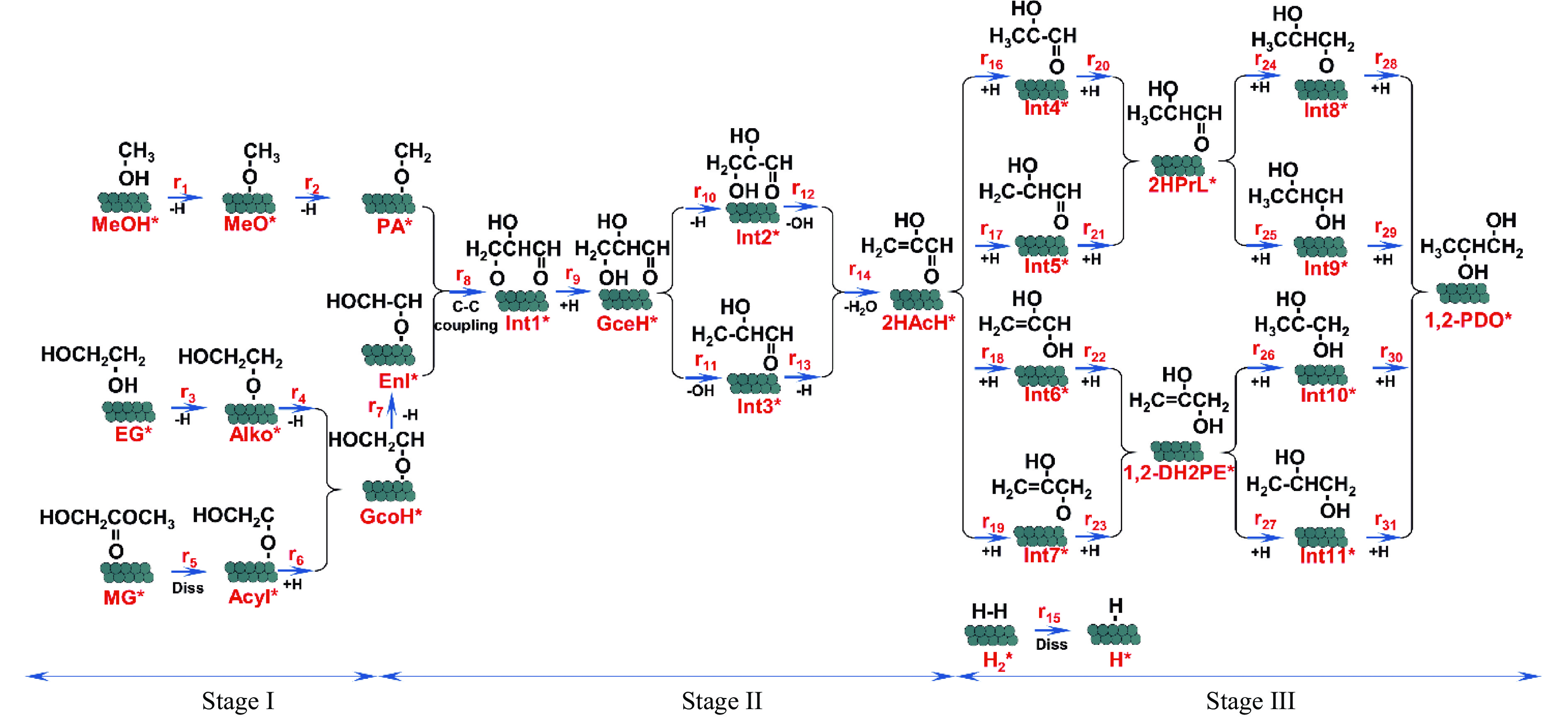
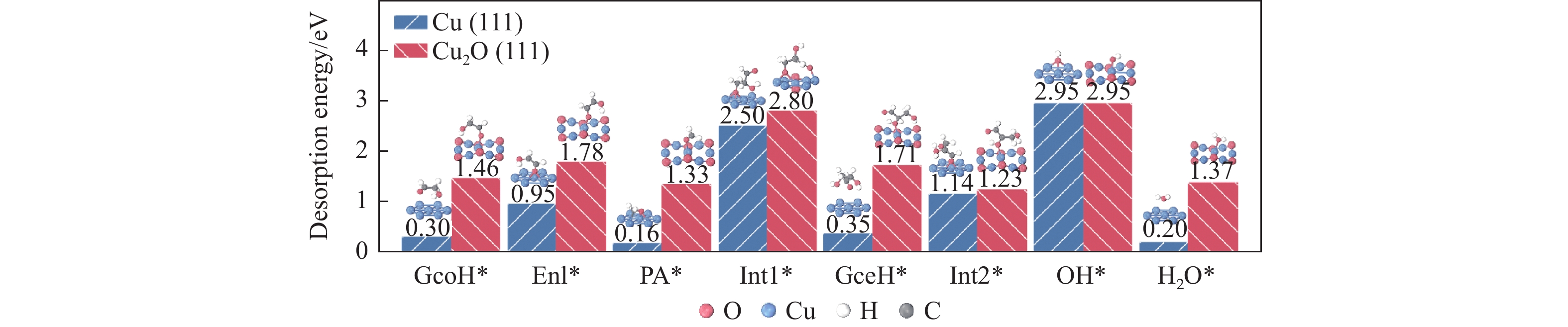
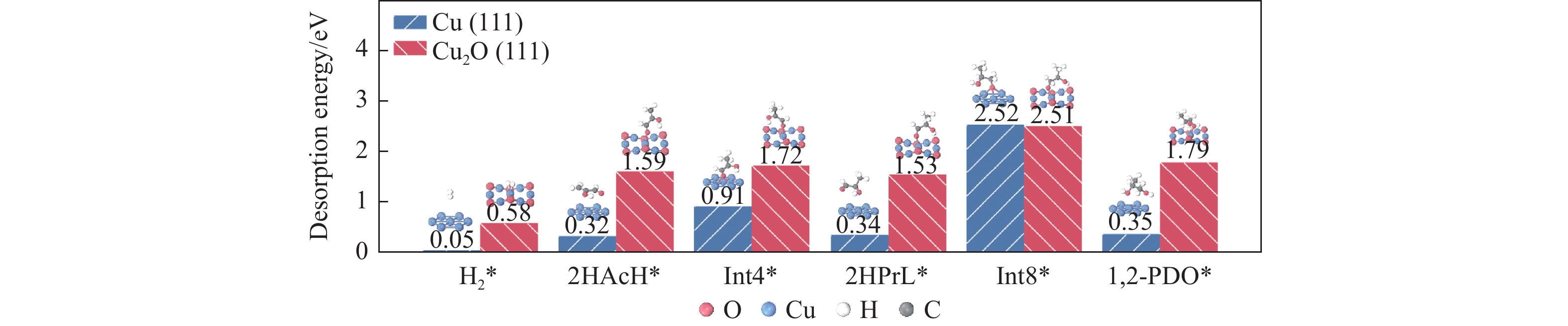
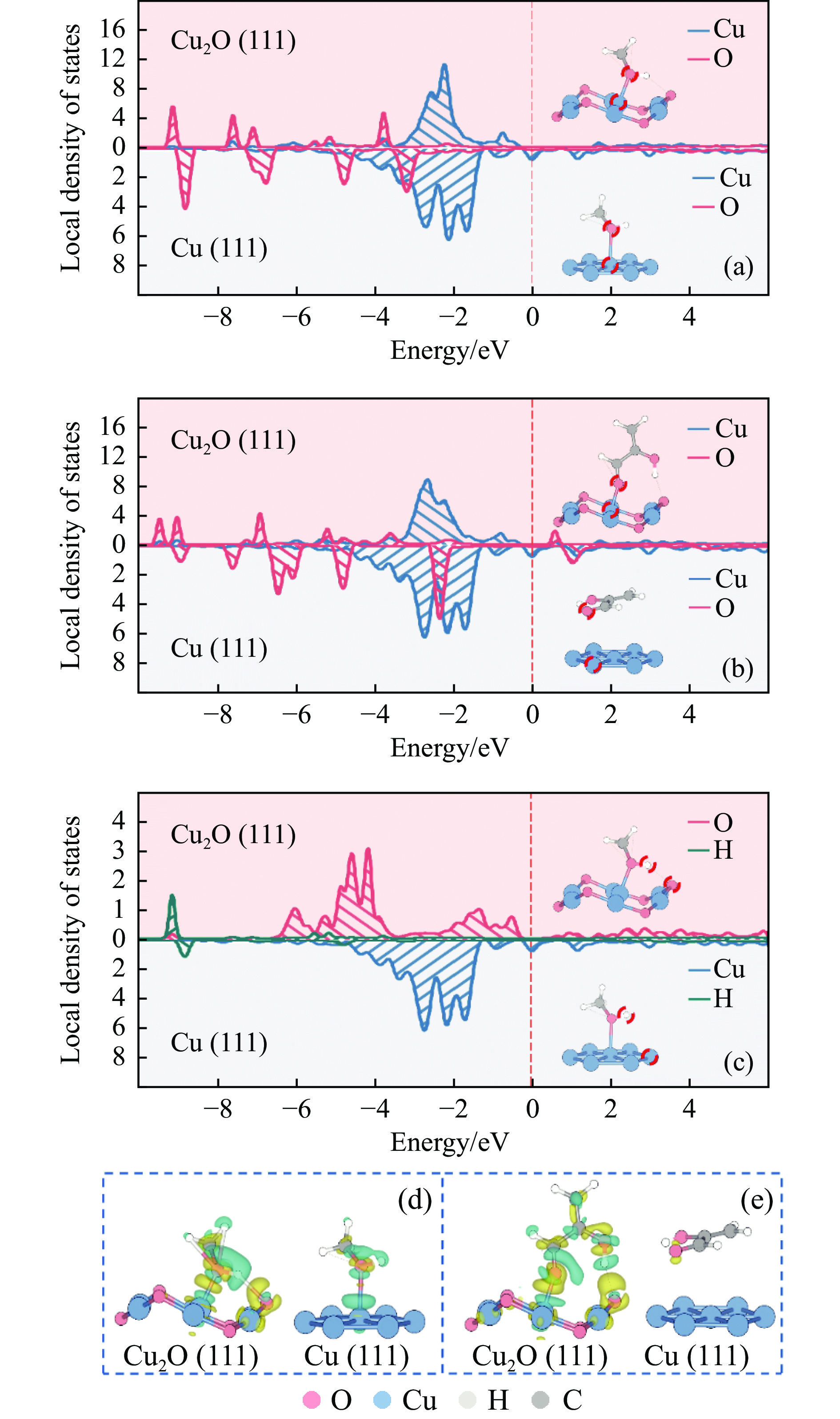
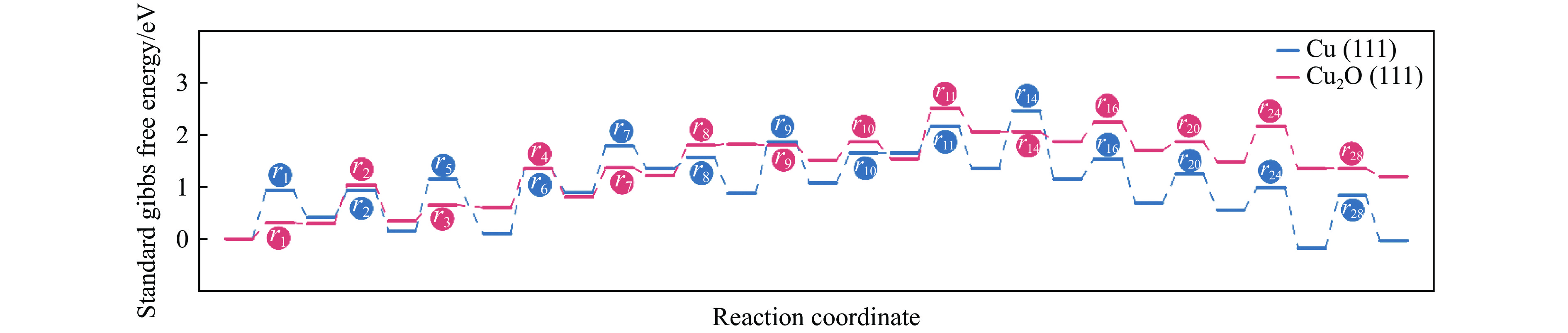
利用密度泛函理论对Cu(111)及Cu2O(111)表面上草酸二甲酯加氢副产物1,2-丙二醇(1,2-PDO)的生成机理进行了探究,计算了两种表面上1,2-PDO生成的不同反应路径基元步骤的热力学数据以及所涉及物种的吸附行为,进行了局域态密度以及差分电荷密度分析,阐明了铜催化剂的主要活性位点及1,2-PDO生成的主要路径。结果表明,1,2-PDO主要由乙二醇和甲醇于Cu2O(111)表面通过Guerbet醇缩合反应生成,具体包括醇脱氢、羟醛缩合以及不饱和醛加氢三个过程。Cu2O(111)表面${\rm{Cu}}_{{\rm{us}}}^{+} $及${\rm{O}}_{{\rm{suf}}}^-$位点形成的Lewis酸碱对能够促进反应物、产物及反应中间体的吸附且对于1,2-PDO生成过程的整体催化活性更高。Cu2O(111)表面的${\rm{O}}_{{\rm{suf}}}^- $位点是醇类脱氢生成醛、羟醛缩合过程中生成烯醇物种以及不饱和醛类中间体加氢的主要活性中心,而C−C偶联反应则发生在${\rm{Cu}}_{{\rm{us}}}^{+} $金属位点上。论文研究结果可为铜催化剂设计和改性以及草酸酯加氢工艺的优化提供理论指导。
利用密度泛函理论对Cu(111)及Cu2O(111)表面上草酸二甲酯加氢副产物1,2-丙二醇(1,2-PDO)的生成机理进行了探究,计算了两种表面上1,2-PDO生成的不同反应路径基元步骤的热力学数据以及所涉及物种的吸附行为,进行了局域态密度以及差分电荷密度分析,阐明了铜催化剂的主要活性位点及1,2-PDO生成的主要路径。结果表明,1,2-PDO主要由乙二醇和甲醇于Cu2O(111)表面通过Guerbet醇缩合反应生成,具体包括醇脱氢、羟醛缩合以及不饱和醛加氢三个过程。Cu2O(111)表面${\rm{Cu}}_{{\rm{us}}}^{+} $及${\rm{O}}_{{\rm{suf}}}^-$位点形成的Lewis酸碱对能够促进反应物、产物及反应中间体的吸附且对于1,2-PDO生成过程的整体催化活性更高。Cu2O(111)表面的${\rm{O}}_{{\rm{suf}}}^- $位点是醇类脱氢生成醛、羟醛缩合过程中生成烯醇物种以及不饱和醛类中间体加氢的主要活性中心,而C−C偶联反应则发生在${\rm{Cu}}_{{\rm{us}}}^{+} $金属位点上。论文研究结果可为铜催化剂设计和改性以及草酸酯加氢工艺的优化提供理论指导。


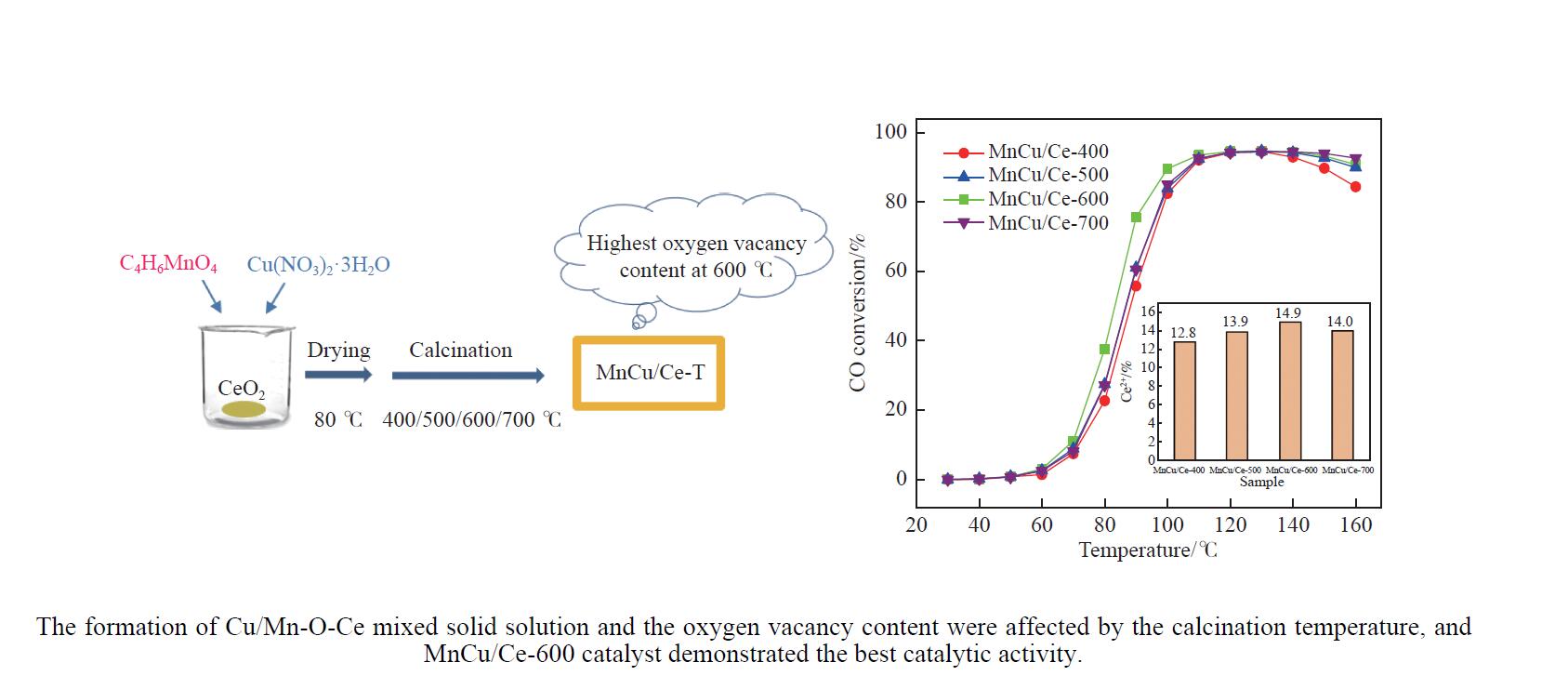
2024, 52(4): 565-576.
doi: 10.19906/j.cnki.JFCT.2023080
摘要:
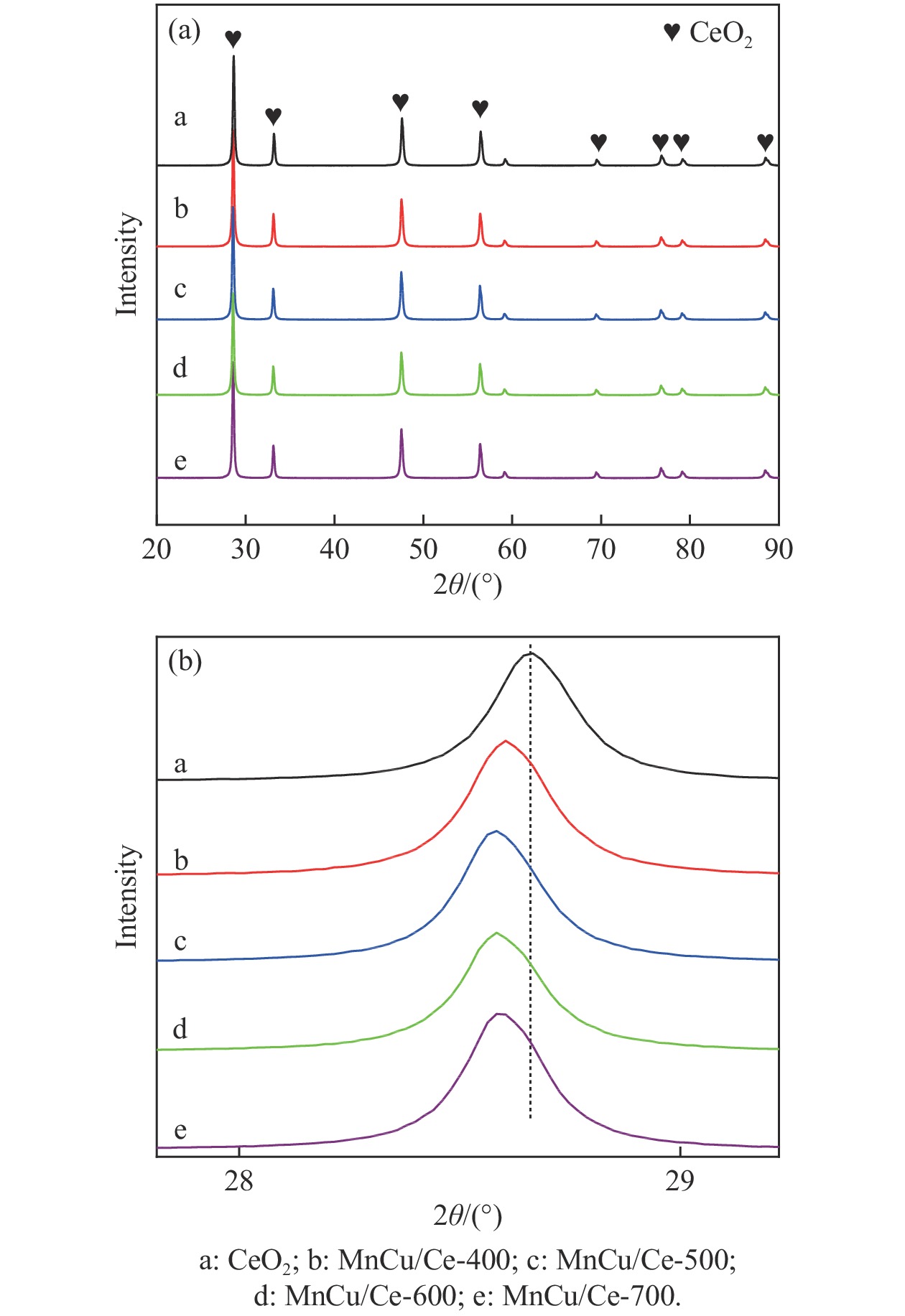
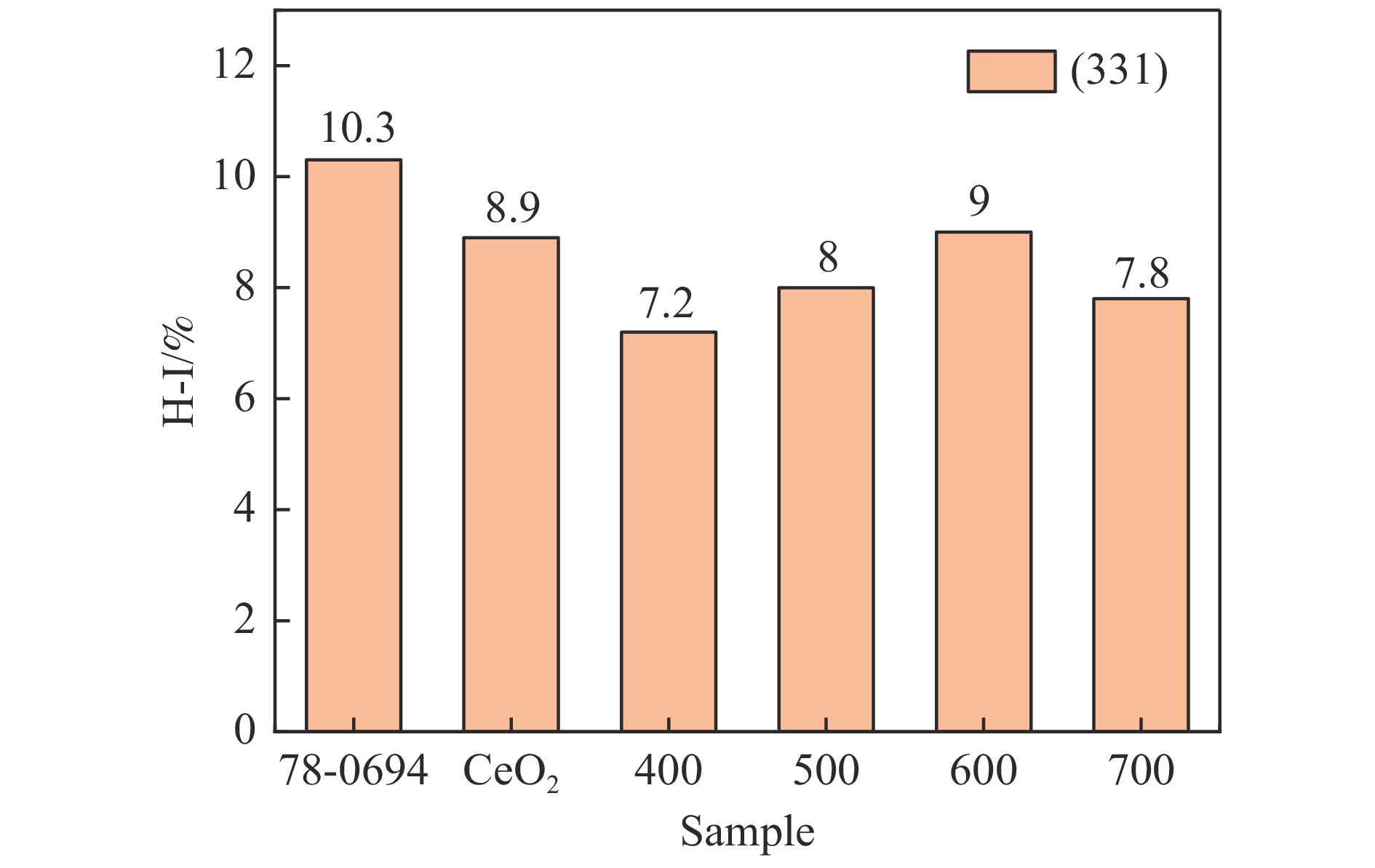
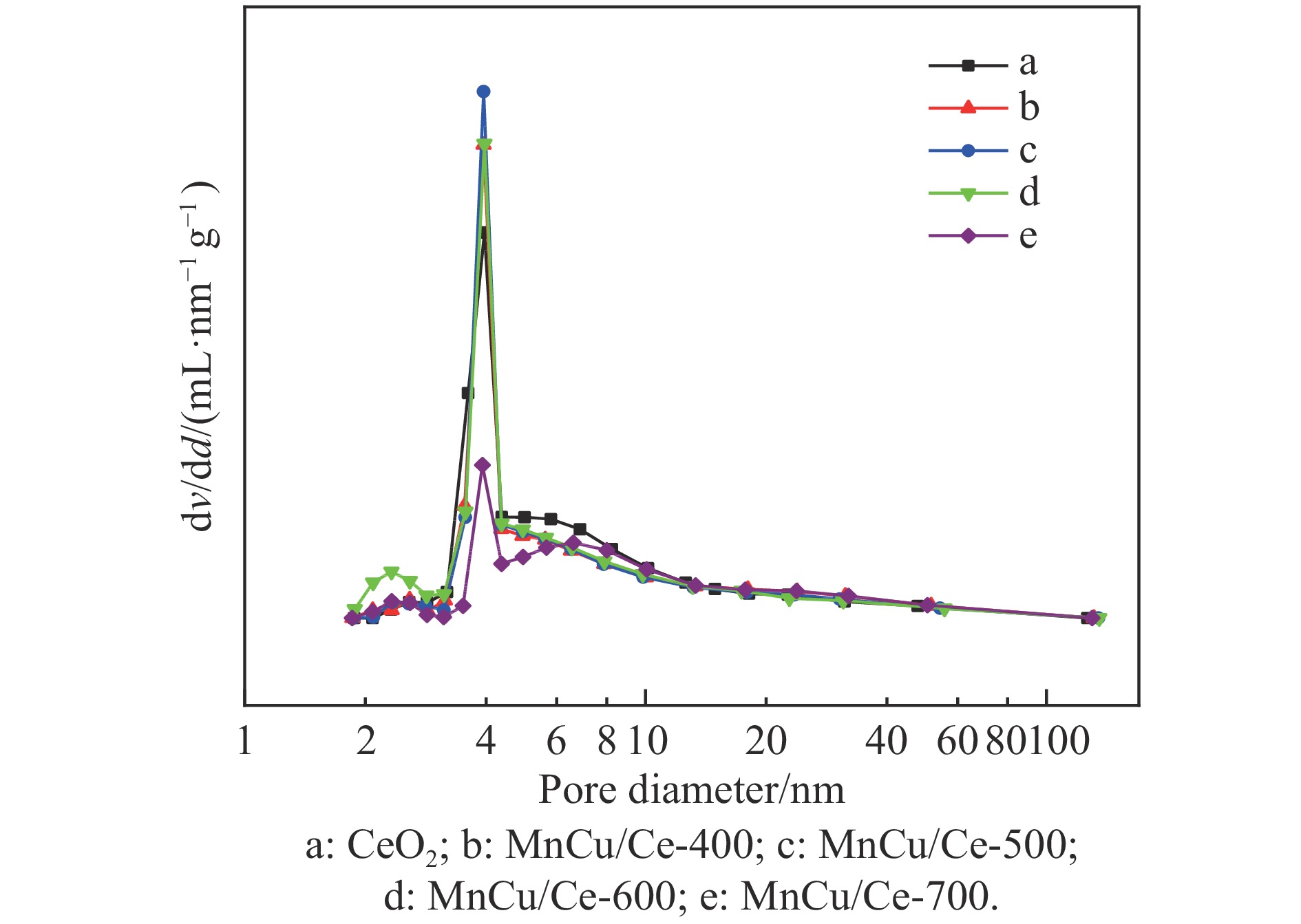
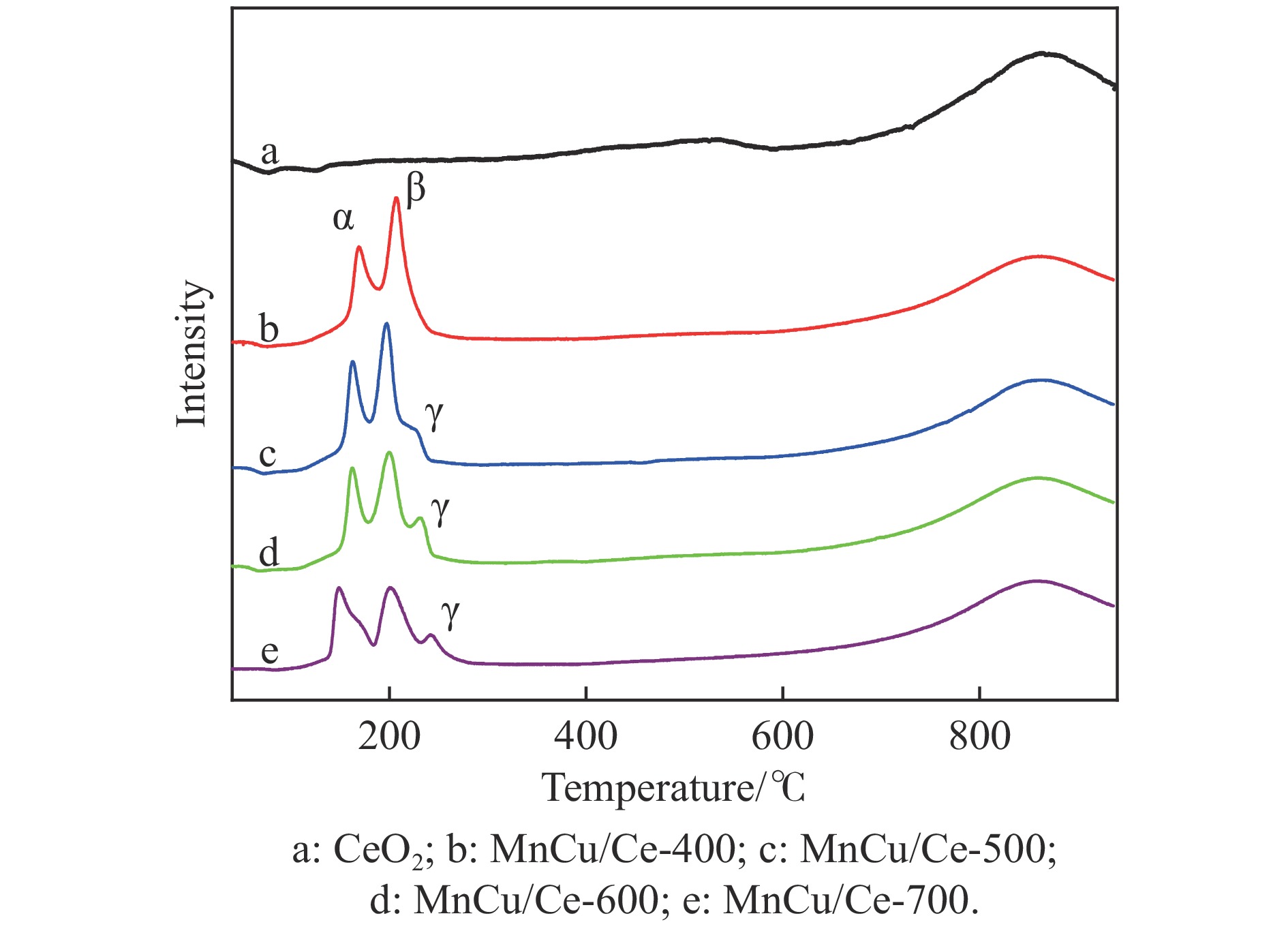
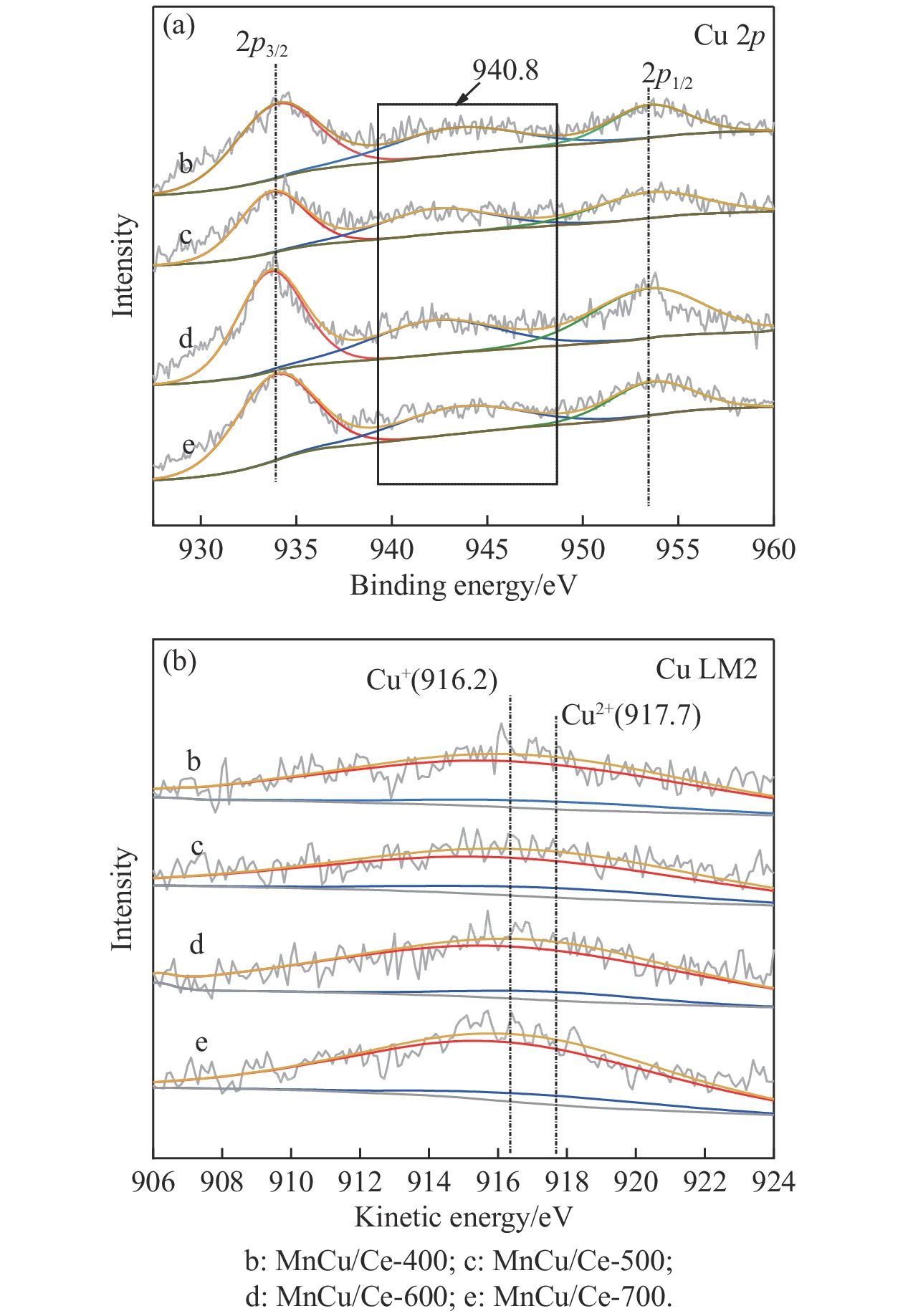
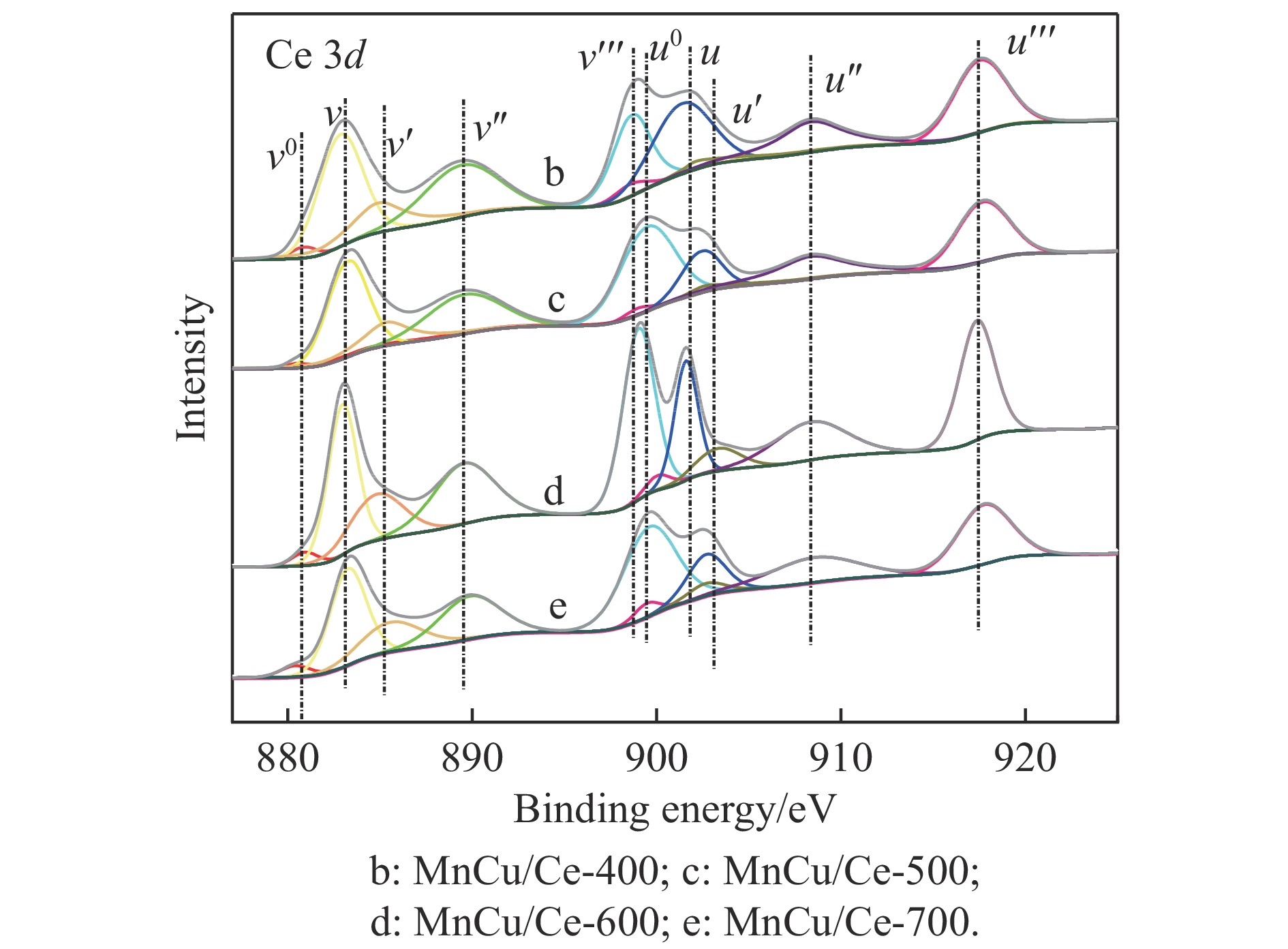
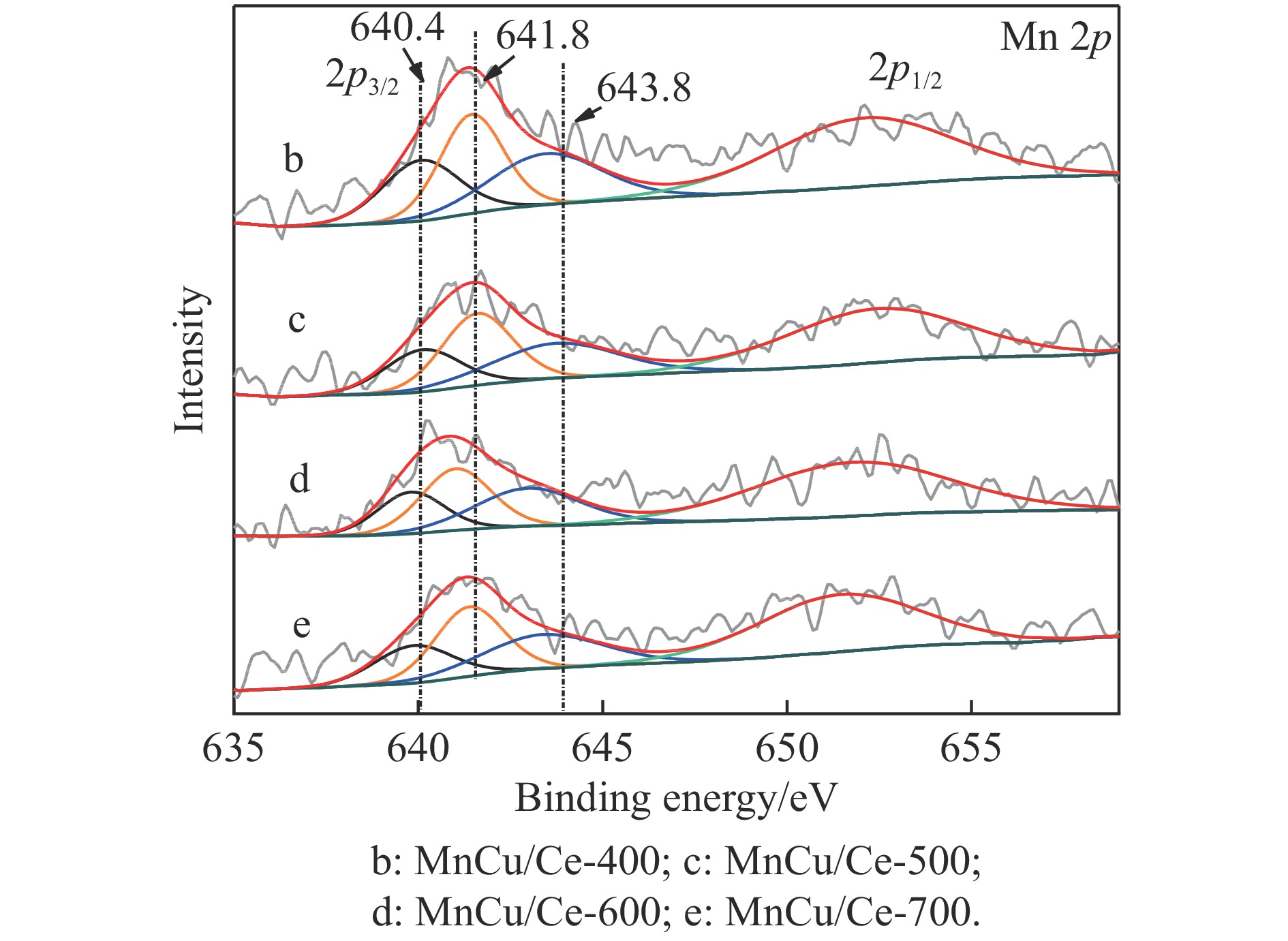
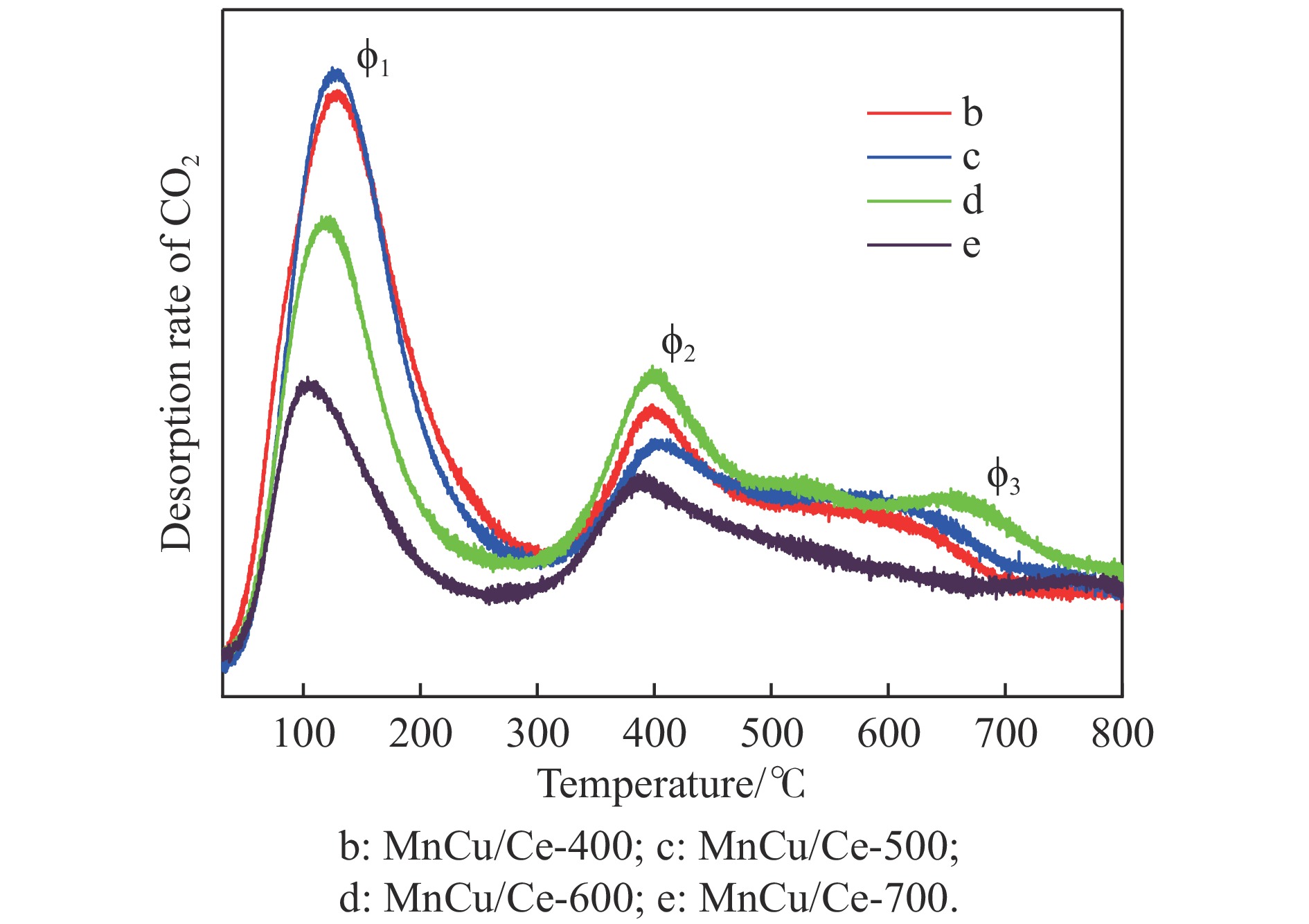
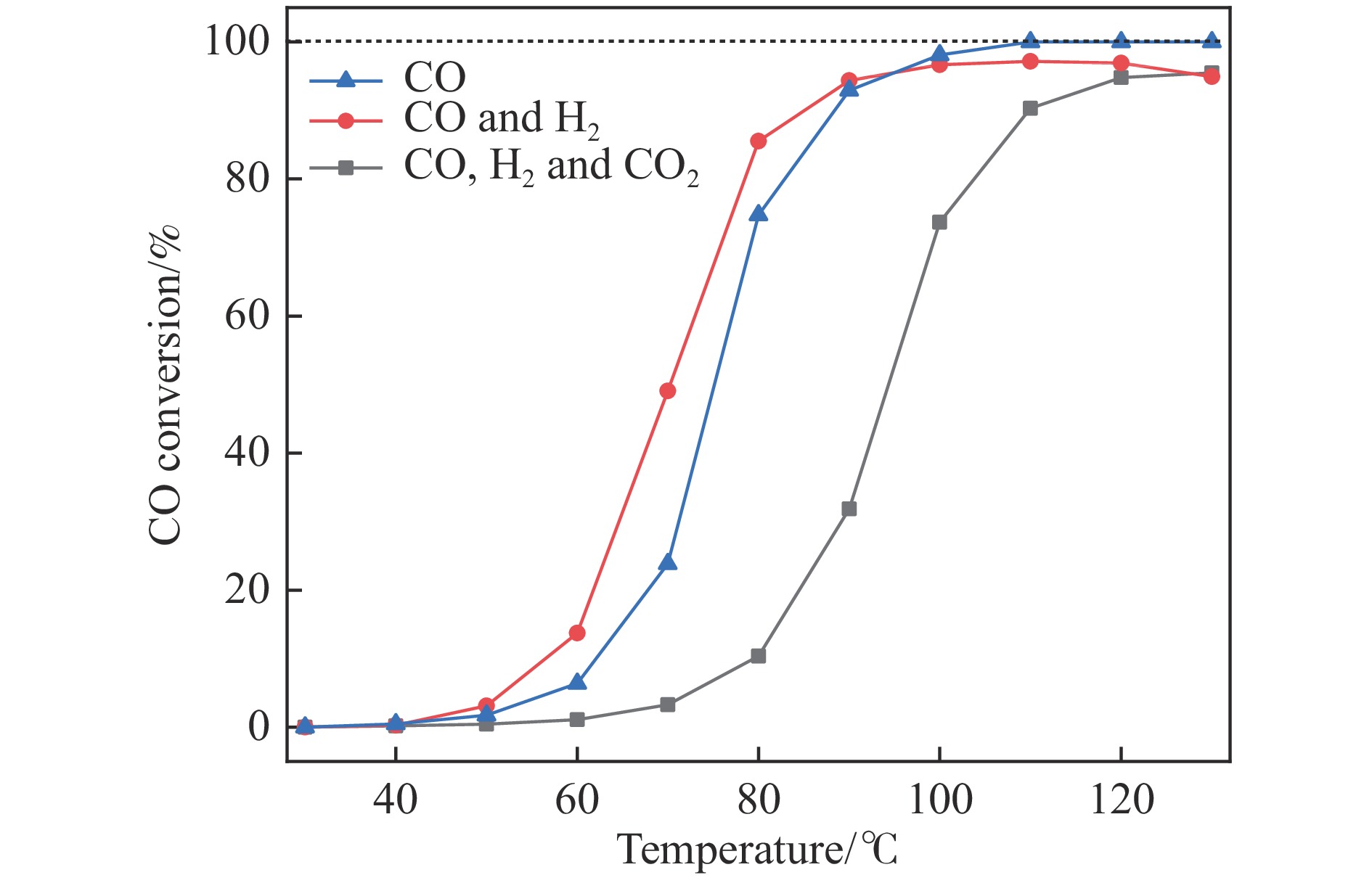
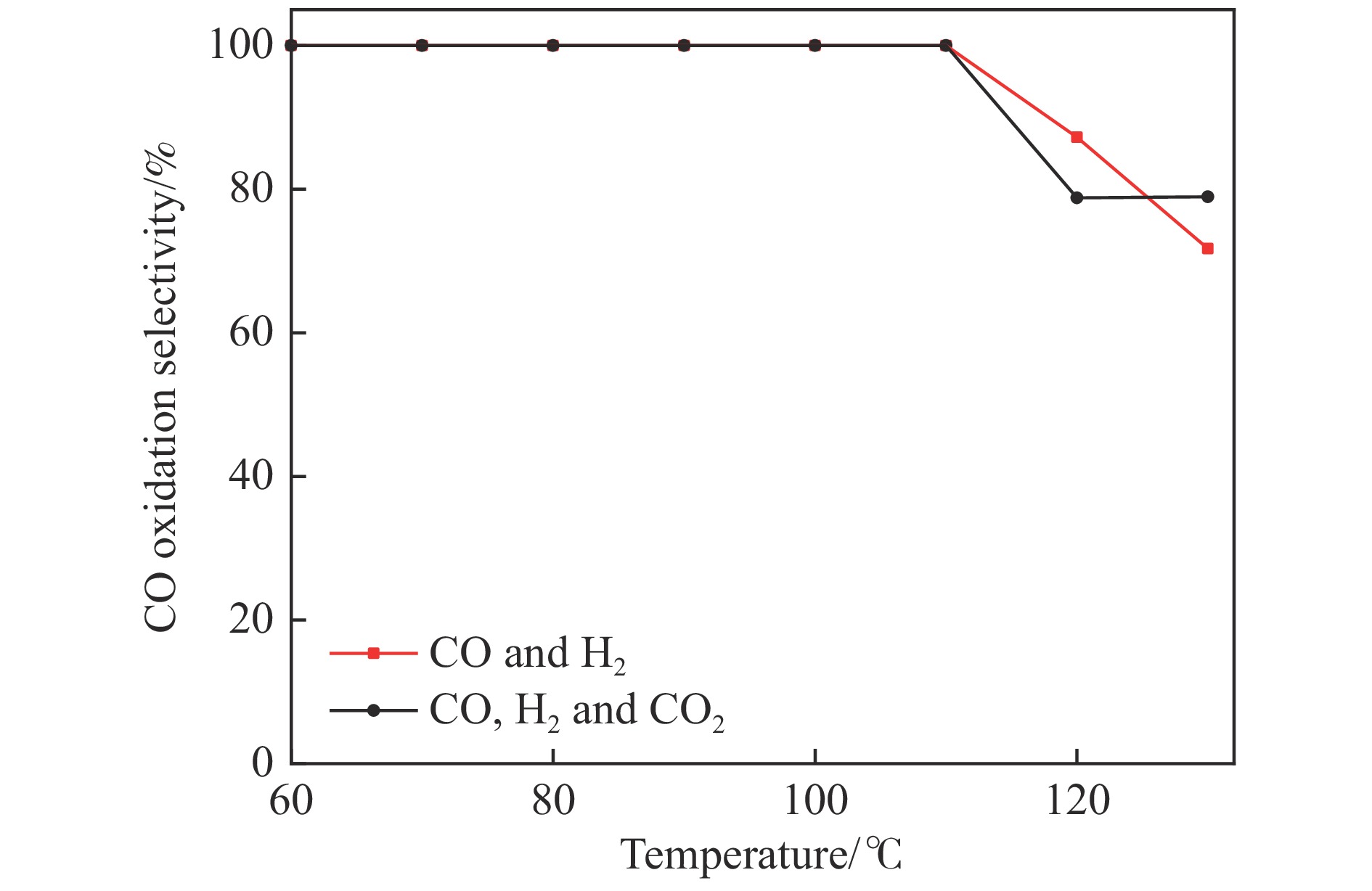
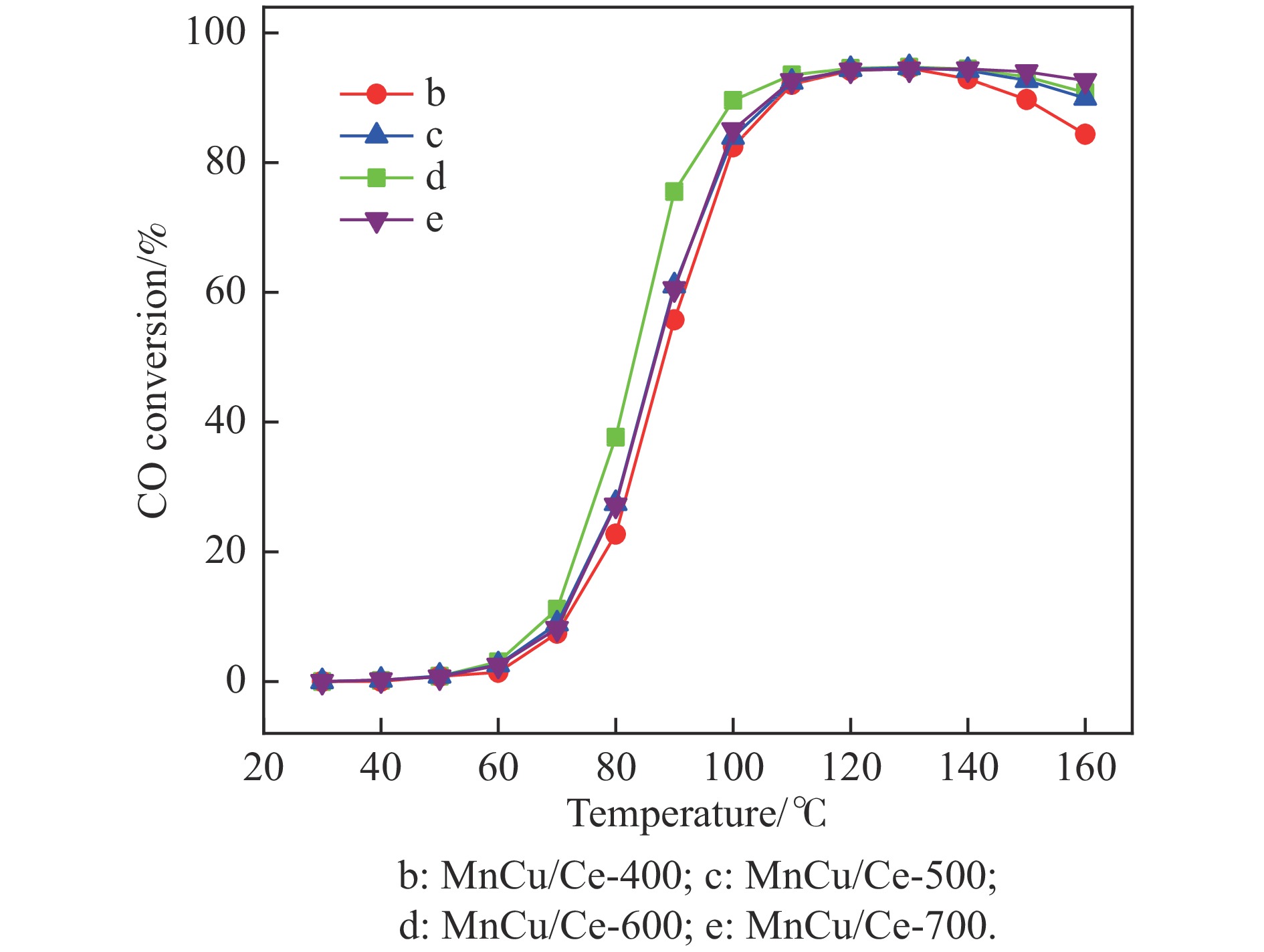
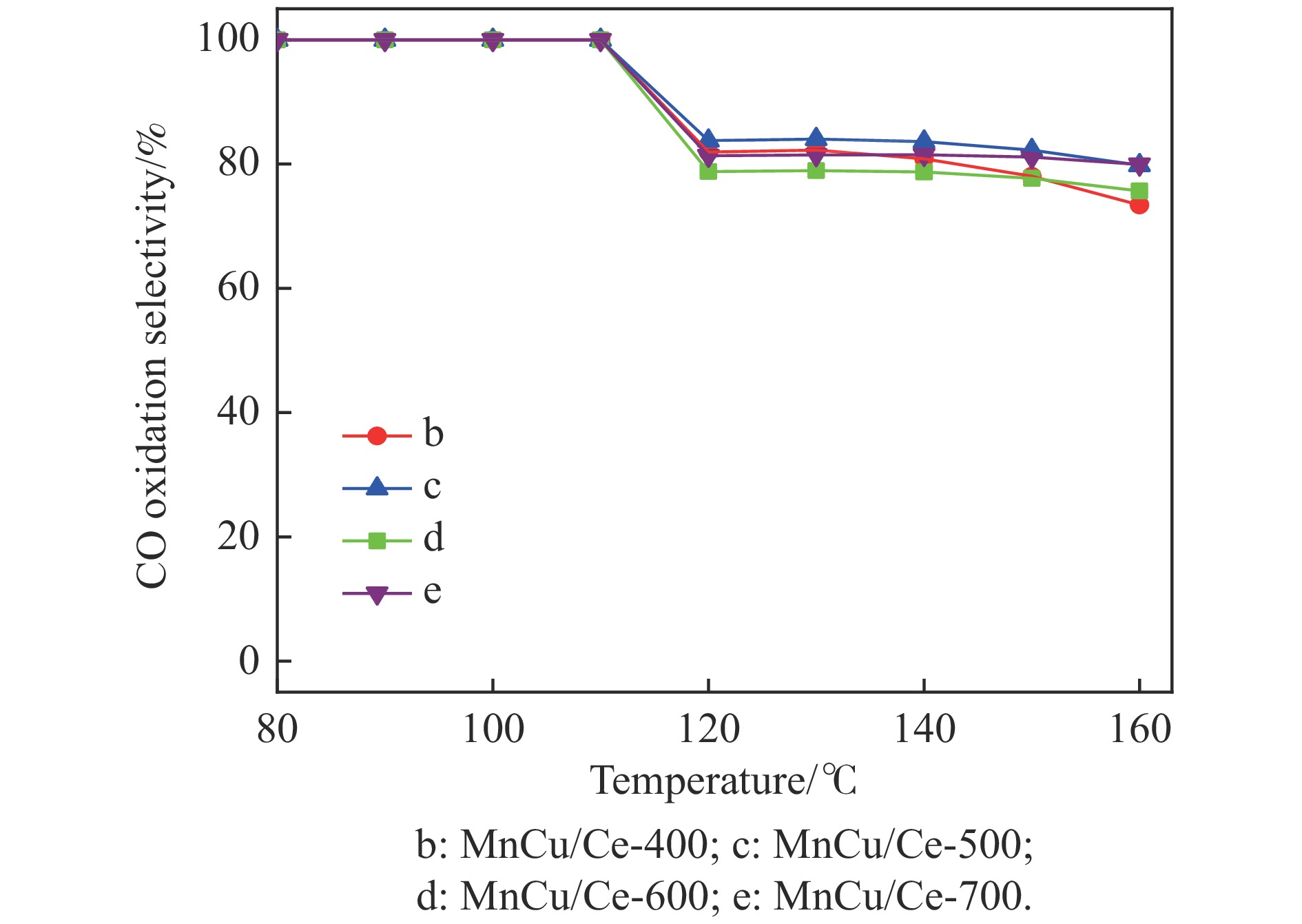
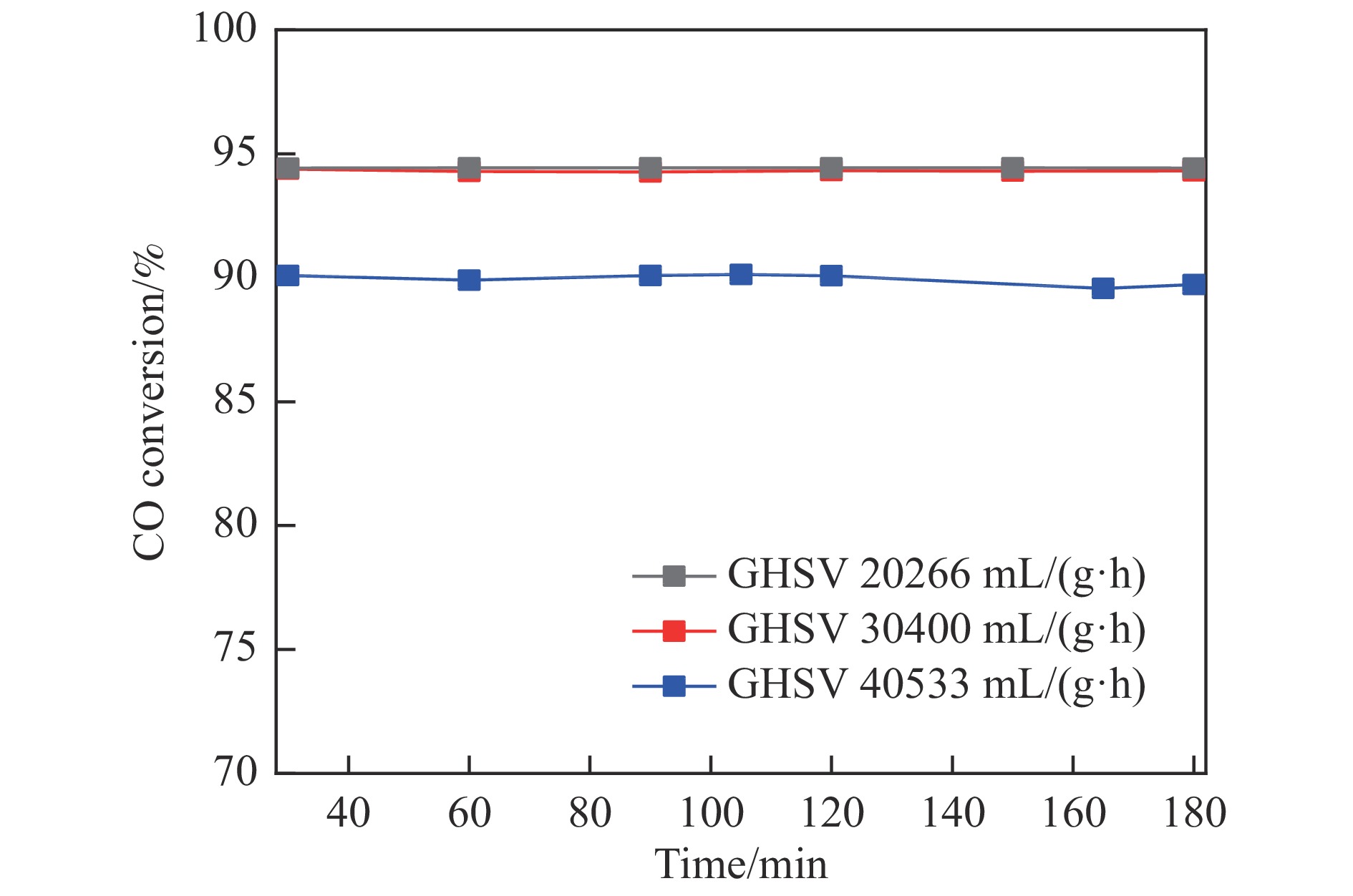
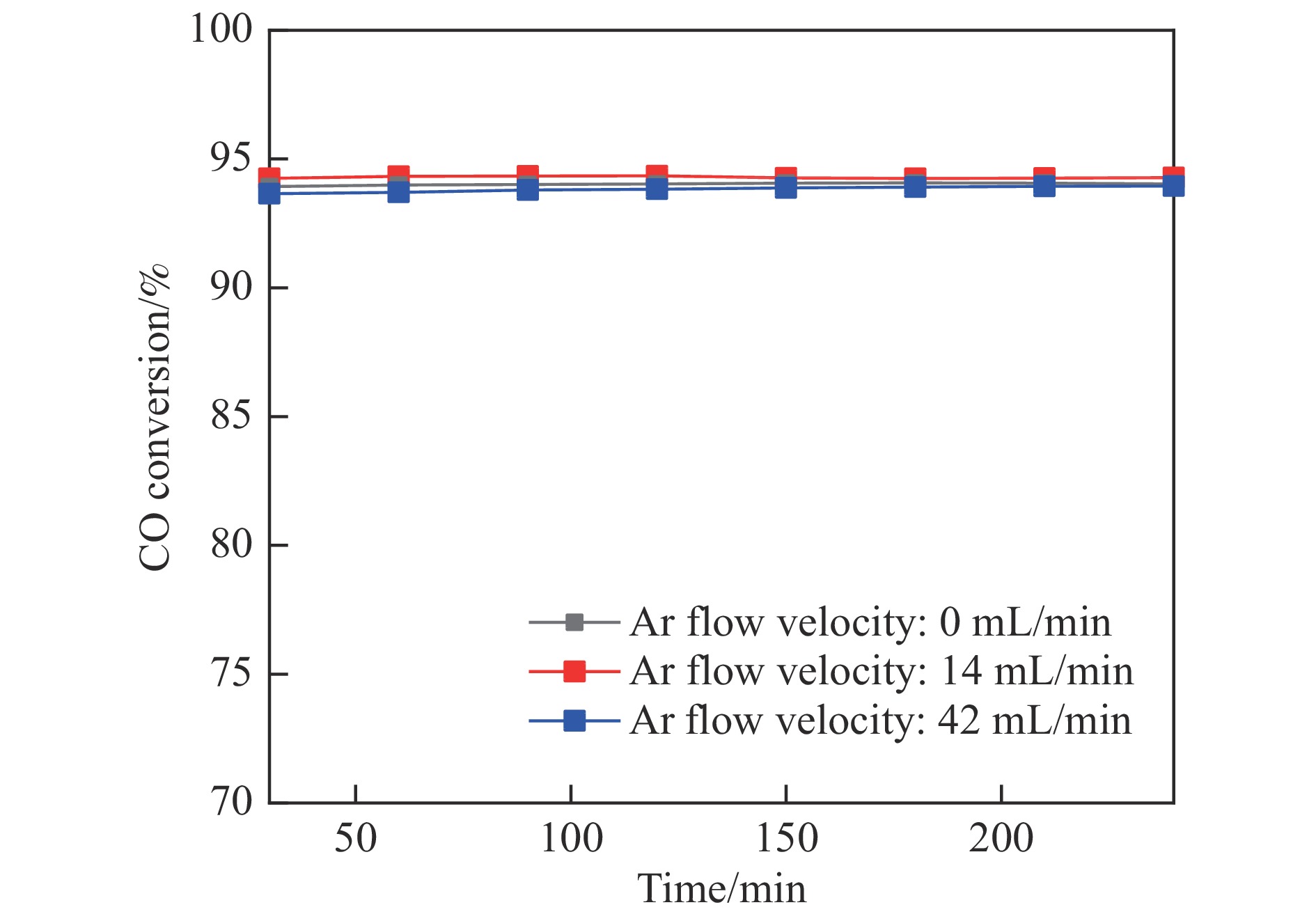
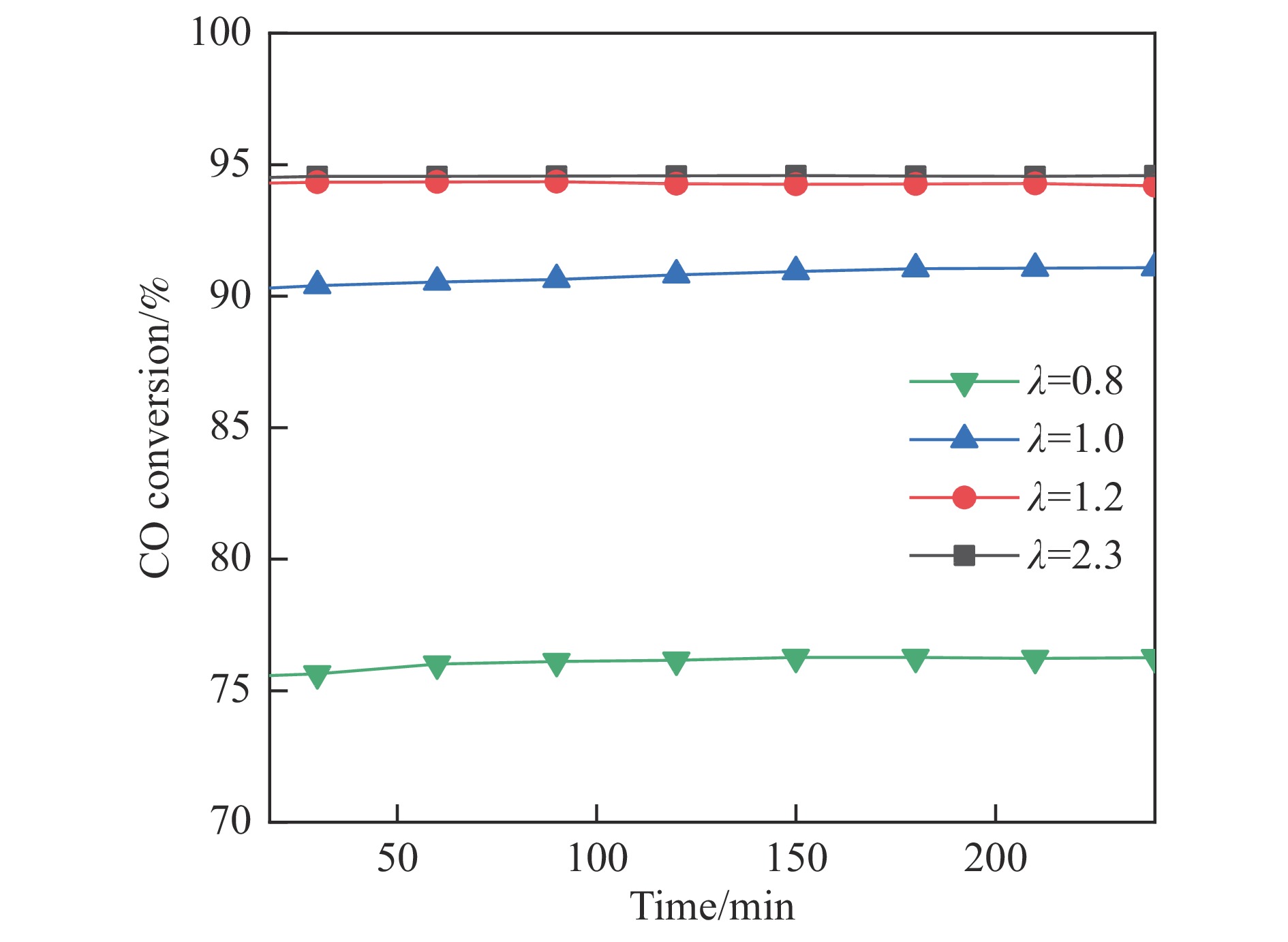
采用共浸渍法制备较低Cu含量的MnCu/Ce催化剂,通过XRD、BET、H2-TPR、XPS和CO2-TPD等表征手段对催化剂进行表征,考察催化剂焙烧温度对催化剂结构、性质及其在含有CO2的富氢气氛下对CO优先氧化性能的影响。结果表明,MnCu/Ce催化剂均有Cu/Mn-O-Ce固溶体形成,其中,在焙烧温度600 ℃制备的催化剂中,Mn与Cu、Ce之间相互作用较强,形成较多三元氧化物固溶体,氧空位/Ce3+含量高,具备良好的CO-Prox活性。此外,对反应条件的考察发现,添加不同分压Ar对催化剂的CO-Prox活性影响较小,气体空速和氧过量系数对催化剂活性影响较大,且反应原料气中CO2的存在对CO-Prox反应有负面影响。氧过量系数为1.2、空速范围为20266−30400 mL/(g·h)时,CO转化率最高,达到94.7%。
采用共浸渍法制备较低Cu含量的MnCu/Ce催化剂,通过XRD、BET、H2-TPR、XPS和CO2-TPD等表征手段对催化剂进行表征,考察催化剂焙烧温度对催化剂结构、性质及其在含有CO2的富氢气氛下对CO优先氧化性能的影响。结果表明,MnCu/Ce催化剂均有Cu/Mn-O-Ce固溶体形成,其中,在焙烧温度600 ℃制备的催化剂中,Mn与Cu、Ce之间相互作用较强,形成较多三元氧化物固溶体,氧空位/Ce3+含量高,具备良好的CO-Prox活性。此外,对反应条件的考察发现,添加不同分压Ar对催化剂的CO-Prox活性影响较小,气体空速和氧过量系数对催化剂活性影响较大,且反应原料气中CO2的存在对CO-Prox反应有负面影响。氧过量系数为1.2、空速范围为20266−30400 mL/(g·h)时,CO转化率最高,达到94.7%。


摘要:
气流床气化过程中产生的细渣含碳量很高,目前多以填埋的方式进行处理,将细渣用于循环流化床锅炉掺烧有望为细渣处理提供有利的技术。本研究选用宁东能源化工基地典型气化工艺GE、OMB及GSP产生的气化细渣为研究对象,利用物理吸附仪、激光拉曼及热重分析仪等仪器,系统研究了气化细渣中残炭的结构特征与燃烧特性。结果表明,原始气化细渣中的物质可分为黏结球形颗粒、多孔不规则颗粒与孤立的大球形颗粒,而酸洗后的气化细渣多以疏松细小的颗粒和多孔不规则块状颗粒存在;细渣中残炭的孔径尺寸主要分布在4−8 nm,且比表面积与残炭的活性位点大小顺序均为:GE > OMB > GSP;GE渣中残炭结构有序度最低,无定形炭结构最多,GSP则相反;GE渣中残炭燃烧速率最快,主要是由于GE渣中残炭有较大的比表面积、较多的无定形炭结构及较高的的活性位点,且GE渣中残炭的综合燃烧指数为5.26 × 10−7%2/(min2·℃3)。
气流床气化过程中产生的细渣含碳量很高,目前多以填埋的方式进行处理,将细渣用于循环流化床锅炉掺烧有望为细渣处理提供有利的技术。本研究选用宁东能源化工基地典型气化工艺GE、OMB及GSP产生的气化细渣为研究对象,利用物理吸附仪、激光拉曼及热重分析仪等仪器,系统研究了气化细渣中残炭的结构特征与燃烧特性。结果表明,原始气化细渣中的物质可分为黏结球形颗粒、多孔不规则颗粒与孤立的大球形颗粒,而酸洗后的气化细渣多以疏松细小的颗粒和多孔不规则块状颗粒存在;细渣中残炭的孔径尺寸主要分布在4−8 nm,且比表面积与残炭的活性位点大小顺序均为:GE > OMB > GSP;GE渣中残炭结构有序度最低,无定形炭结构最多,GSP则相反;GE渣中残炭燃烧速率最快,主要是由于GE渣中残炭有较大的比表面积、较多的无定形炭结构及较高的的活性位点,且GE渣中残炭的综合燃烧指数为5.26 × 10−7%2/(min2·℃3)。
摘要:
利用CaO基吸附剂直接从高温烟气中捕集CO2因成本低、吸附性能好等优点成为CCUS(碳捕集、利用与封存)的重要技术之一。但CaO基吸附剂在碳酸化/煅烧吸脱附循环过程中存在易烧结的问题,导致其吸附性能急剧下降。本研究针对CaO基吸附剂全面总结了其吸附CO2的动力学、热力学及烧结机理,并重点综述了世界各国研究者在CaO基吸附剂抗烧结改性方面所做的研究,指出了各种方法的优点及其局限性。结果表明,水合作用改性可使吸附剂崩塌而获得更大的比表面积;酸溶液改性会在制备过程产生更多的气体和小分子物质提高吸附剂孔隙率;掺杂改性可以促进CaO对CO2的吸附和扩散,还可作为骨架分离CaO颗粒。经比较,掺杂改性工艺简单、性能好,是比较有前景的改性方法,以含钙固废制备抗烧结改性吸附剂是发展方向。
利用CaO基吸附剂直接从高温烟气中捕集CO2因成本低、吸附性能好等优点成为CCUS(碳捕集、利用与封存)的重要技术之一。但CaO基吸附剂在碳酸化/煅烧吸脱附循环过程中存在易烧结的问题,导致其吸附性能急剧下降。本研究针对CaO基吸附剂全面总结了其吸附CO2的动力学、热力学及烧结机理,并重点综述了世界各国研究者在CaO基吸附剂抗烧结改性方面所做的研究,指出了各种方法的优点及其局限性。结果表明,水合作用改性可使吸附剂崩塌而获得更大的比表面积;酸溶液改性会在制备过程产生更多的气体和小分子物质提高吸附剂孔隙率;掺杂改性可以促进CaO对CO2的吸附和扩散,还可作为骨架分离CaO颗粒。经比较,掺杂改性工艺简单、性能好,是比较有前景的改性方法,以含钙固废制备抗烧结改性吸附剂是发展方向。
摘要:
Waste gasification has the potential to contribute to China’s transition towards carbon neutrality and zero waste cities via the recirculation of waste as secondary carbon feedstock for the production of chemicals with lower/and or zero carbon footprint, green hydrogen with zero carbon footprint and CO2-neutral synthetic liquid fuels. With China’s significant coal gasification capacity and associated experiences and expertise, Coal-to-X could act as a bridge to Waste-to-X for carbon intensive sectors such as the waste management, chemical production and mobility sectors. To illustrate the opportunities in these areas, this article presented highlights from dynamic global developments in waste gasification, focusing on pioneering industrial developments in Germany between 1980−2000’s as well as current international developments. Lessons learnt from previous and current waste gasification project deployment are shared and enabled the identification of problems which will have to be addressed in the transition from coal gasification towards mono-waste gasification technologies. Additionally, a qualitative evaluation of gasification technologies pointed to the strengths and weaknesses of fixed-bed, fluidized-bed and entrained-flow gasification principles in their application for waste gasification.
Waste gasification has the potential to contribute to China’s transition towards carbon neutrality and zero waste cities via the recirculation of waste as secondary carbon feedstock for the production of chemicals with lower/and or zero carbon footprint, green hydrogen with zero carbon footprint and CO2-neutral synthetic liquid fuels. With China’s significant coal gasification capacity and associated experiences and expertise, Coal-to-X could act as a bridge to Waste-to-X for carbon intensive sectors such as the waste management, chemical production and mobility sectors. To illustrate the opportunities in these areas, this article presented highlights from dynamic global developments in waste gasification, focusing on pioneering industrial developments in Germany between 1980−2000’s as well as current international developments. Lessons learnt from previous and current waste gasification project deployment are shared and enabled the identification of problems which will have to be addressed in the transition from coal gasification towards mono-waste gasification technologies. Additionally, a qualitative evaluation of gasification technologies pointed to the strengths and weaknesses of fixed-bed, fluidized-bed and entrained-flow gasification principles in their application for waste gasification.
摘要:
在NH3选择性催化还原(NH3-SCR)反应中,由于具有宽温度窗口和良好的水热稳定性,金属负载型分子筛是具有广泛应用潜力的脱硝催化剂。本文综述了Cu基和Fe基分子筛催化剂在NH3-SCR领域的研究进展,总结了催化剂的结构特征和NH3-SCR性能指标,并对相应的金属活性位点和反应机理进行了归纳。此外,系统介绍了密度泛函理论(DFT)计算在NH3-SCR反应机理中的应用及反应动力学的研究方法,并对比了不同催化剂体系下的表观动力学参数,为进一步研究金属负载型分子筛催化剂的NH3-SCR反应机理提供方法与思路。
在NH3选择性催化还原(NH3-SCR)反应中,由于具有宽温度窗口和良好的水热稳定性,金属负载型分子筛是具有广泛应用潜力的脱硝催化剂。本文综述了Cu基和Fe基分子筛催化剂在NH3-SCR领域的研究进展,总结了催化剂的结构特征和NH3-SCR性能指标,并对相应的金属活性位点和反应机理进行了归纳。此外,系统介绍了密度泛函理论(DFT)计算在NH3-SCR反应机理中的应用及反应动力学的研究方法,并对比了不同催化剂体系下的表观动力学参数,为进一步研究金属负载型分子筛催化剂的NH3-SCR反应机理提供方法与思路。
摘要:
The chemical and mineralogical characteristics of fly ash from a municipal solid waste incineration (MSWI) in China and the influence of processing parameters on heavy metals removal during leaching were investigated in this work. The fly ash particles had complex surface structure with limited specific surface area. The alkali chloride and metal salts in MSWI fly ash showed evidently impact on leaching efficiency. Metal leachability was related to their properties and speciation in fly ash. Water-soluble salts such as KCl, NaCl and CaCl2 in fly ash were easily washed out. In this study, removal efficiency by water washing was achieved to 93.1% for Cl, 41.4% for Na, 48.5% for K and 24.8% for Ca, respectively. Mineralogical analysis also revealed change of fly ash mineral phases and specification distribution after water washing. Under liquid to solid ratio of 40∶1 L/kg and treatment time of 120 min, the leaching process achieved high dropping yields of toxicity characteristic leaching procedure (TCLP) concentrations for Cu, Zn Cd and Pb (80%−100%), moderate dropping yields for As (30%−80%) and relatively low dropping yields of Ni (< 30%). In addition, heavy metals such as Pb and Zn in fly ash with twice water washing treatment at a low liquid-solid ratio could reach lower TCLP concentrations. The result indicated that the combination process of twice water washing and one acid washing could significantly reduce the environmental risk of MSWI fly ash.
The chemical and mineralogical characteristics of fly ash from a municipal solid waste incineration (MSWI) in China and the influence of processing parameters on heavy metals removal during leaching were investigated in this work. The fly ash particles had complex surface structure with limited specific surface area. The alkali chloride and metal salts in MSWI fly ash showed evidently impact on leaching efficiency. Metal leachability was related to their properties and speciation in fly ash. Water-soluble salts such as KCl, NaCl and CaCl2 in fly ash were easily washed out. In this study, removal efficiency by water washing was achieved to 93.1% for Cl, 41.4% for Na, 48.5% for K and 24.8% for Ca, respectively. Mineralogical analysis also revealed change of fly ash mineral phases and specification distribution after water washing. Under liquid to solid ratio of 40∶1 L/kg and treatment time of 120 min, the leaching process achieved high dropping yields of toxicity characteristic leaching procedure (TCLP) concentrations for Cu, Zn Cd and Pb (80%−100%), moderate dropping yields for As (30%−80%) and relatively low dropping yields of Ni (< 30%). In addition, heavy metals such as Pb and Zn in fly ash with twice water washing treatment at a low liquid-solid ratio could reach lower TCLP concentrations. The result indicated that the combination process of twice water washing and one acid washing could significantly reduce the environmental risk of MSWI fly ash.
摘要:
考察了Pb对Mn-Ce/TiO2低温选择性催化还原(SCR)脱硝活性的影响,并对Pb中毒的催化剂进行了再生;结合氮吸附、SEM、XRD、FT-IR、H2-TPR和NH3-TPD等表征结果,研究了Mn-Ce/TiO2催化剂Pb中毒和再生活性恢复的原因。结果表明,Pb对Mn-Ce/TiO2催化剂脱硝活性有明显的抑制作用;当Pb的含量为11%时,Mn-Ce/TiO2催化剂在180 ℃下的脱硝效率从原来100%下降至44%。Pb在Mn-Ce/TiO2中的掺杂使得催化剂的比表面积以及活性组分Mn4+和Ce3+的含量降低,影响了氧化还原循环反应(Mn4+ + Ce3+ ↔ Mn3+ + Ce4+)的进行;此外,Pb的加入破坏了催化剂的酸性位点,阻碍了催化剂对NH3的吸附和活化。经硝酸再生后的Mn-Ce/TiO2催化剂的脱硝活性几乎完全恢复,在80–150 ℃下其脱硝活性甚至超过新鲜未中毒的催化剂,其原因主要在于硝酸再生能恢复催化剂的氧化还原能力、增大比表面积、并形成新的酸位点。
考察了Pb对Mn-Ce/TiO2低温选择性催化还原(SCR)脱硝活性的影响,并对Pb中毒的催化剂进行了再生;结合氮吸附、SEM、XRD、FT-IR、H2-TPR和NH3-TPD等表征结果,研究了Mn-Ce/TiO2催化剂Pb中毒和再生活性恢复的原因。结果表明,Pb对Mn-Ce/TiO2催化剂脱硝活性有明显的抑制作用;当Pb的含量为11%时,Mn-Ce/TiO2催化剂在180 ℃下的脱硝效率从原来100%下降至44%。Pb在Mn-Ce/TiO2中的掺杂使得催化剂的比表面积以及活性组分Mn4+和Ce3+的含量降低,影响了氧化还原循环反应(Mn4+ + Ce3+ ↔ Mn3+ + Ce4+)的进行;此外,Pb的加入破坏了催化剂的酸性位点,阻碍了催化剂对NH3的吸附和活化。经硝酸再生后的Mn-Ce/TiO2催化剂的脱硝活性几乎完全恢复,在80–150 ℃下其脱硝活性甚至超过新鲜未中毒的催化剂,其原因主要在于硝酸再生能恢复催化剂的氧化还原能力、增大比表面积、并形成新的酸位点。
摘要:
采用溶胶-凝胶-超临界干燥法、水热法及共沉淀法分别合成了氧化铈气凝胶(CeO2-A)、纳米棒(CeO2-R)和纳米片(CeO2-F)。考察了不同形貌氧化铈的催化燃烧甲苯性能,通过多种方法分析表征了氧化铈样品的微观结构,讨论了不同方法制得的CeO2形貌结构对催化性能的影响。结果表明,CeO2-R和CeO2-F比表面积较低,并且仅暴露(111)晶面,催化燃烧甲苯活性较低。CeO2-A具有高比表面积和丰富的孔道结构,有利于反应物分子的吸附,而且同时暴露(100)和(111)两种活性晶面,增加了氧空位浓度(Osur/Olatt = 0.25)。此外,CeO2-A由于表面晶格氧移动性较强,有利于Ce3+/Ce4+氧化还原的循环,加快甲苯深度氧化反应的进行。因此,CeO2-A具有更加优异的催化燃烧甲苯活性,t50和t90分别为223 和239 ℃,这主要归因于其大比表面积、高暴露活性晶面以及强晶格氧迁移性。
采用溶胶-凝胶-超临界干燥法、水热法及共沉淀法分别合成了氧化铈气凝胶(CeO2-A)、纳米棒(CeO2-R)和纳米片(CeO2-F)。考察了不同形貌氧化铈的催化燃烧甲苯性能,通过多种方法分析表征了氧化铈样品的微观结构,讨论了不同方法制得的CeO2形貌结构对催化性能的影响。结果表明,CeO2-R和CeO2-F比表面积较低,并且仅暴露(111)晶面,催化燃烧甲苯活性较低。CeO2-A具有高比表面积和丰富的孔道结构,有利于反应物分子的吸附,而且同时暴露(100)和(111)两种活性晶面,增加了氧空位浓度(Osur/Olatt = 0.25)。此外,CeO2-A由于表面晶格氧移动性较强,有利于Ce3+/Ce4+氧化还原的循环,加快甲苯深度氧化反应的进行。因此,CeO2-A具有更加优异的催化燃烧甲苯活性,t50和t90分别为223 和239 ℃,这主要归因于其大比表面积、高暴露活性晶面以及强晶格氧迁移性。
摘要:
本文采用浸渍法制备了Nb改性的V2O5-WO3/TiO2催化剂,研究了脱硝反应中Nb负载量对催化剂SO2氧化活性的影响。结果表明,在350 °C下,Nb2O5负载量为2%的Nb2O5-V2O5-WO3/TiO2催化剂上的SO2氧化率最低(0.6%),而同时NOx 的转化率仍能达到95%。采用TGA、氮吸附、XRD、H2-TPR、CO2-TPD、XPS和in- situ DRIFTS等对催化剂进行了表征分析,结果显示,Nb改性后V2O5-WO3/TiO2催化剂的晶体结构没有发生明显改变,但是其比表面积小幅度下降,有助于减少对SO2的吸附;同时,改性后催化剂表面的吸附氧含量下降,氧化还原性能也稍微减弱,这有利于降低其对SO2的氧化活性。in-situ DRIFTS结果表明,Nb改性后的Nb-V2O5-WO3/TiO2催化剂反应过程中表面中间产物VOSO4的含量明显下降,从而减少了SO3的生成量。
本文采用浸渍法制备了Nb改性的V2O5-WO3/TiO2催化剂,研究了脱硝反应中Nb负载量对催化剂SO2氧化活性的影响。结果表明,在350 °C下,Nb2O5负载量为2%的Nb2O5-V2O5-WO3/TiO2催化剂上的SO2氧化率最低(0.6%),而同时NOx 的转化率仍能达到95%。采用TGA、氮吸附、XRD、H2-TPR、CO2-TPD、XPS和in- situ DRIFTS等对催化剂进行了表征分析,结果显示,Nb改性后V2O5-WO3/TiO2催化剂的晶体结构没有发生明显改变,但是其比表面积小幅度下降,有助于减少对SO2的吸附;同时,改性后催化剂表面的吸附氧含量下降,氧化还原性能也稍微减弱,这有利于降低其对SO2的氧化活性。in-situ DRIFTS结果表明,Nb改性后的Nb-V2O5-WO3/TiO2催化剂反应过程中表面中间产物VOSO4的含量明显下降,从而减少了SO3的生成量。
摘要:
纤维素的热解技术是一种非常有应用前景的高值转化技术。本综述系统地介绍了纤维素的基础特性,深入讨论了纤维素热解机制、研究方法、催化剂类型及其他影响纤维素热解产物分布的因素。其中,不同类型催化剂的添加和反应装置结构的设计优化可以显著提高纤维素热解转化效率,改善产物种类分布和提高特定高值化学品的选择性,从而有效地提高纤维素热解产物的资源、能源化利用价值。最后,对纤维素热解未来技术研究的发展方向进行了展望。
纤维素的热解技术是一种非常有应用前景的高值转化技术。本综述系统地介绍了纤维素的基础特性,深入讨论了纤维素热解机制、研究方法、催化剂类型及其他影响纤维素热解产物分布的因素。其中,不同类型催化剂的添加和反应装置结构的设计优化可以显著提高纤维素热解转化效率,改善产物种类分布和提高特定高值化学品的选择性,从而有效地提高纤维素热解产物的资源、能源化利用价值。最后,对纤维素热解未来技术研究的发展方向进行了展望。
摘要:
化石资源的大量使用导致CO2的大量排放,带来了严重的环境问题。与此同时,CO2又是一种清洁、无毒的含碳资源。将CO2作为原料,直接转化制备重要化学品,不仅可以减缓温室效应,同时也是一条有效利用含碳资源制备清洁燃料和化学品的新路线。本文概述了近年来关于CO2加氢制备一些烃类化合物(主要包括甲烷、烯烃和芳烃)的相关研究进展;重点分析了CO2加氢制烃类化合物相关过程催化剂的研发状态和对催化反应机理的认识,并对CO2加氢转化利用的未来发展进行了展望。
化石资源的大量使用导致CO2的大量排放,带来了严重的环境问题。与此同时,CO2又是一种清洁、无毒的含碳资源。将CO2作为原料,直接转化制备重要化学品,不仅可以减缓温室效应,同时也是一条有效利用含碳资源制备清洁燃料和化学品的新路线。本文概述了近年来关于CO2加氢制备一些烃类化合物(主要包括甲烷、烯烃和芳烃)的相关研究进展;重点分析了CO2加氢制烃类化合物相关过程催化剂的研发状态和对催化反应机理的认识,并对CO2加氢转化利用的未来发展进行了展望。
摘要:
以生物质(葡萄糖、蔗糖、淀粉)为模板剂,通过水热合成法制备具有核壳结构的CuO-ZnO-Al2O3@Al2O3复合催化剂,该催化剂以甲醇合成催化剂CuO-ZnO-Al2O3为核,甲醇脱水催化剂Al2O3为壳.SEM-EDS对催化剂核壳结构的表征发现,通过改变水热合成温度和合成时间可以调变催化剂中Al2O3壳层的厚度.将该复合催化剂用于合成气直接制备二甲醚的反应,在空速1 500 mL/(h·gcat)、温度260 ℃、压力5.0 MPa的条件下,CO转化率和二甲醚选择性分别达到35.2%和61.1%.
以生物质(葡萄糖、蔗糖、淀粉)为模板剂,通过水热合成法制备具有核壳结构的CuO-ZnO-Al2O3@Al2O3复合催化剂,该催化剂以甲醇合成催化剂CuO-ZnO-Al2O3为核,甲醇脱水催化剂Al2O3为壳.SEM-EDS对催化剂核壳结构的表征发现,通过改变水热合成温度和合成时间可以调变催化剂中Al2O3壳层的厚度.将该复合催化剂用于合成气直接制备二甲醚的反应,在空速1 500 mL/(h·gcat)、温度260 ℃、压力5.0 MPa的条件下,CO转化率和二甲醚选择性分别达到35.2%和61.1%.
摘要:
通过10℃/min时的热重分析,对五种不同性质污泥分别在氮气和氧气气氛下的热解和燃烧特性进行了研究。结果表明,污水处理工艺的“好氧+厌氧”、“厌氧+好氧”过程以及污泥的厌氧消化均使污泥中的有机物结构复杂,导致污泥热解时有机物的分解和析出温度升高,且“好氧+厌氧”过程使污泥中有机物结构更复杂;而对污泥的燃烧过程和燃尽点无影响,但使着火温度升高。利用 šatava-šesták 方程对污泥热解、燃烧的反应机理进行了研究。结果表明,五种污泥热解时均呈现为挥发分扩散和随后的化学反应机理函数,而燃烧时则为化学反应和随后的扩散过程。
通过10℃/min时的热重分析,对五种不同性质污泥分别在氮气和氧气气氛下的热解和燃烧特性进行了研究。结果表明,污水处理工艺的“好氧+厌氧”、“厌氧+好氧”过程以及污泥的厌氧消化均使污泥中的有机物结构复杂,导致污泥热解时有机物的分解和析出温度升高,且“好氧+厌氧”过程使污泥中有机物结构更复杂;而对污泥的燃烧过程和燃尽点无影响,但使着火温度升高。利用 šatava-šesták 方程对污泥热解、燃烧的反应机理进行了研究。结果表明,五种污泥热解时均呈现为挥发分扩散和随后的化学反应机理函数,而燃烧时则为化学反应和随后的扩散过程。
