

Theoretical Calculations of Pyridine Adsorption on the Surfaces of Ti, Zr, N Doped Graphene
-
摘要: 采用密度泛函方法,研究了Ti、Zr和N掺杂及本征石墨烯对柴油中典型碱性氮化物吡啶的吸附行为,讨论了相应的吸附能、吸附构型、马利肯电荷转移、差分电荷密度和态密度。结果表明,金属Ti、Zr掺杂能显著增强吡啶在石墨烯表面的吸附能,非金属N掺杂可略微增加吡啶和石墨烯表面间的吸附能。吡啶在不同原子修饰的石墨烯表面的吸附能大小顺序为Ti掺杂石墨烯>Zr掺杂石墨烯>N掺杂石墨烯>本征石墨烯,吡啶可与Ti、Zr掺杂石墨烯发生N-Ti、N-Zr和π-π作用,与N掺杂石墨烯、本征石墨烯发生N-N、C-N和π-π作用。进一步分析发现,吡啶和金属Ti、Zr掺杂石墨烯表面存在明显的电子转移和化学键的形成,而和非金属N掺杂石墨烯及本征石墨烯间并无化学键形成。吡啶与Ti、Zr掺杂石墨烯发生化学吸附,与N掺杂石墨烯、本征石墨烯发生物理吸附。吡啶更稳定的吸附在Ti、Zr掺杂石墨烯表面。Abstract: The removal of nitrides from diesel fuel has important significance for the environment and human health. The adsorption behaviour of Ti, Zr and N-doped and intrinsic graphene on pyridine, a typical basic nitride in diesel fuel, has been investigated by density functional methods in this paper. the corresponding adsorption energy, adsorption configurations, Mulliken charge transfer, differential charge density, and density of states were discussed. The results show that metal Ti and Zr doping can significantly enhance the adsorption energy between pyridine and graphene surfaces, and non-metal N doping can slightly increase the adsorption energy between pyridine and graphene surfaces. The magnitude of the adsorption energy of pyridine on the surface of graphene modified with different atoms was in the order of Ti doped graphene > Zr doped graphene > N doped graphene > intrinsic graphene, Pyridine could undergo N-Ti, N-Zr and π-π interactions with Ti and Zr doped graphene, and N-N, C-N and π-π interactions with N doped graphene and intrinsic graphene. Further analysis reveals that there are obvious electron transfer and chemical bond formation between pyridine and metallic Ti, Zr-doped graphene surfaces, while there is no chemical bond formation with non-metallic N-doped graphene and intrinsic graphene. Chemical adsorption interaction of pyridine with Ti, Zr-doped graphene, physical adsorption interaction with N-doped graphene and intrinsic graphene. Pyridine was more stable adsorption on the surface of Ti Zr-doped graphene.
-
Key words:
- graphene /
- dope /
- adsorption /
- denitrification /
- simulation /
- surface
-
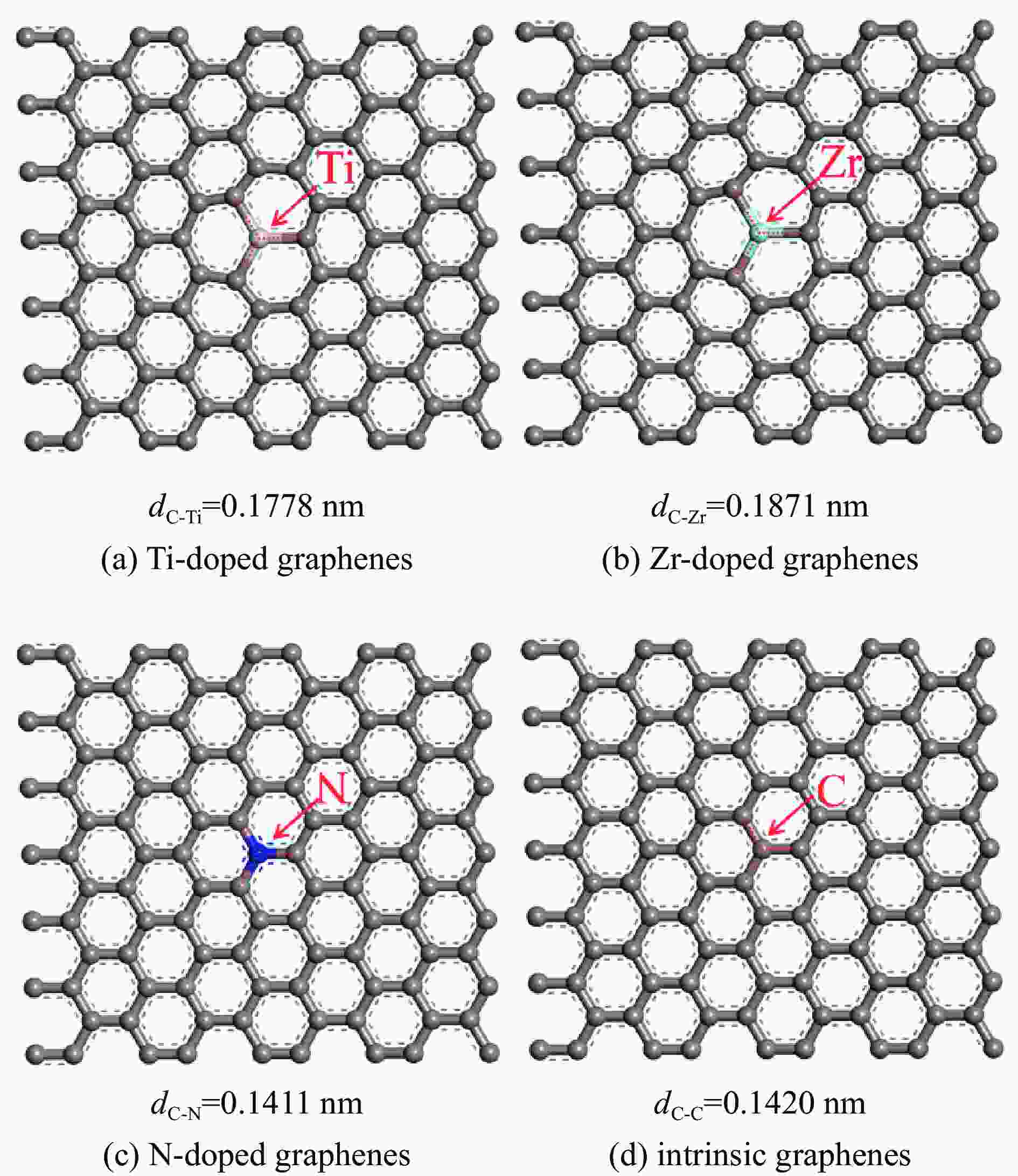
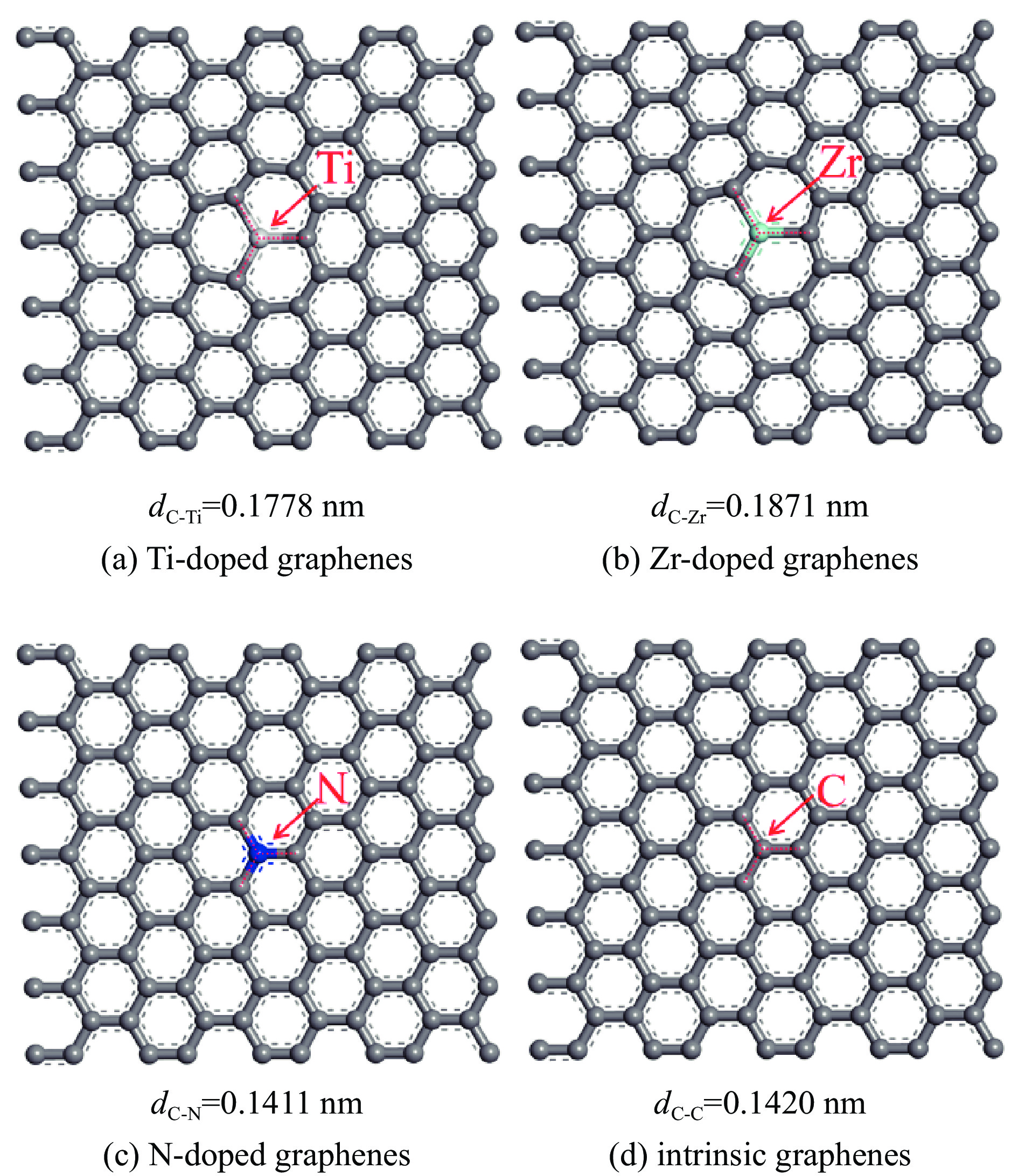
图 1 Ti、Zr、N掺杂及本征石墨烯表面的球棍模型
Figure 1 Ball and stick model of intrinsic and Ti, Zr, N doped graphenes(Titanium, Zirconium, Nitrogen, carbon atoms are represented by incanus, green, blue, grey, respectively)
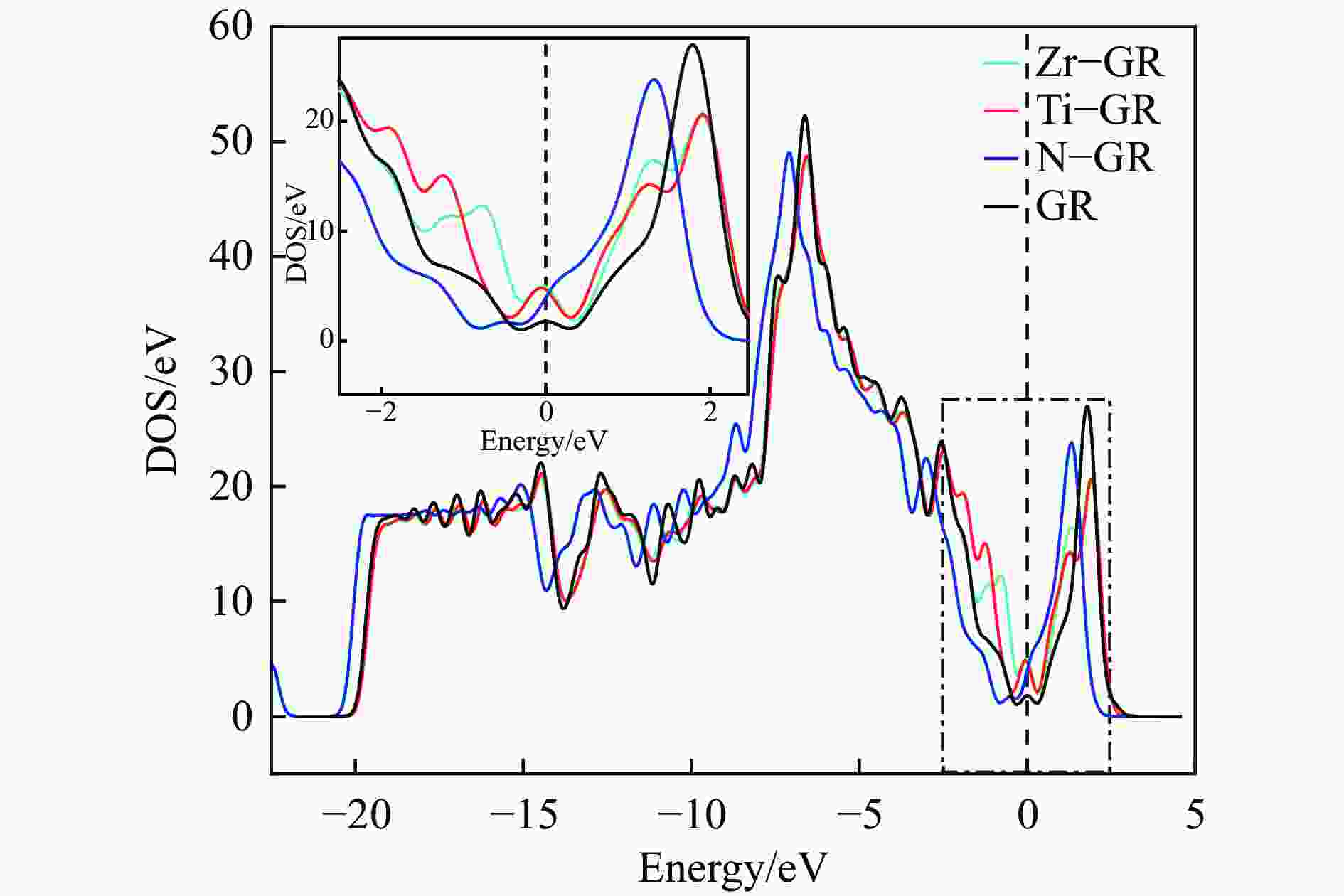
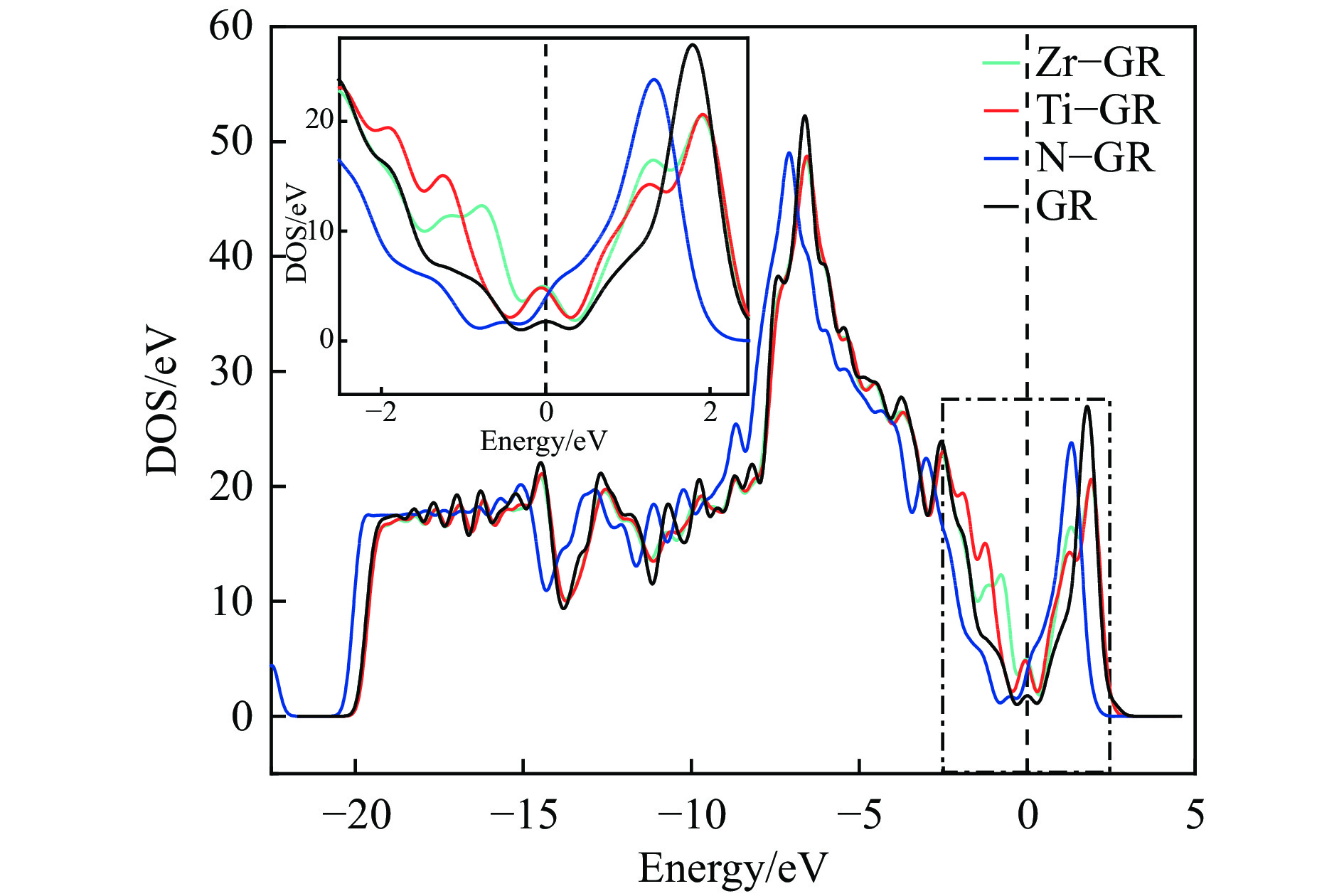
图 2 本征及掺杂石墨烯的态密度
Figure 2 Total density of states of intrinsic and Ti、Zr、N doped graphenes
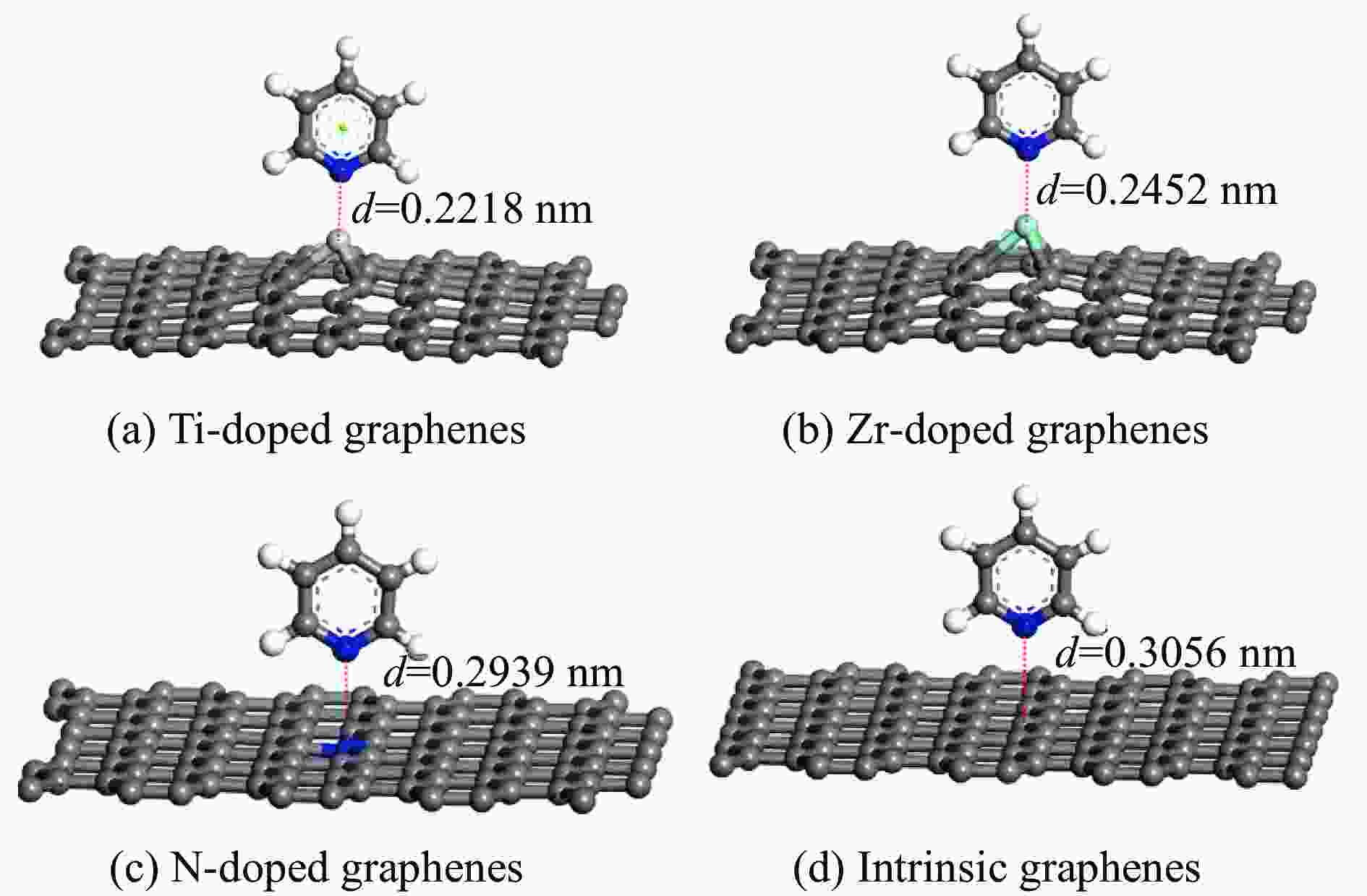
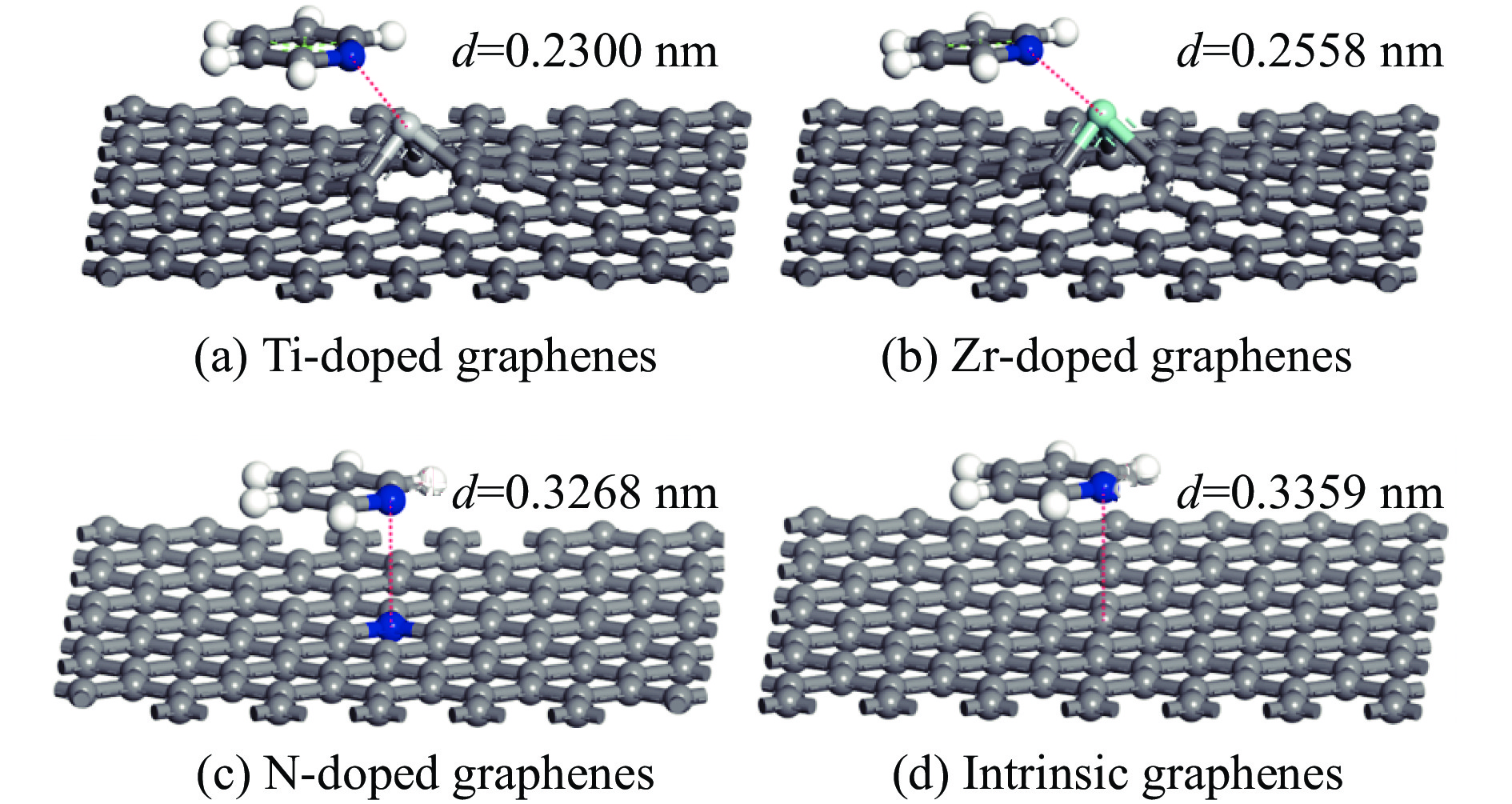
图 3 吡啶垂直吸附在各石墨烯表面的吸附构型
Figure 3 Pyridine vertical adsorption on intrinsic and Ti、Zr、N doped graphenes (Titanium, Zirconium, Nitrogen, carbon and hydrogen atoms are represented by incanus, green, blue, grey and white, respectively)
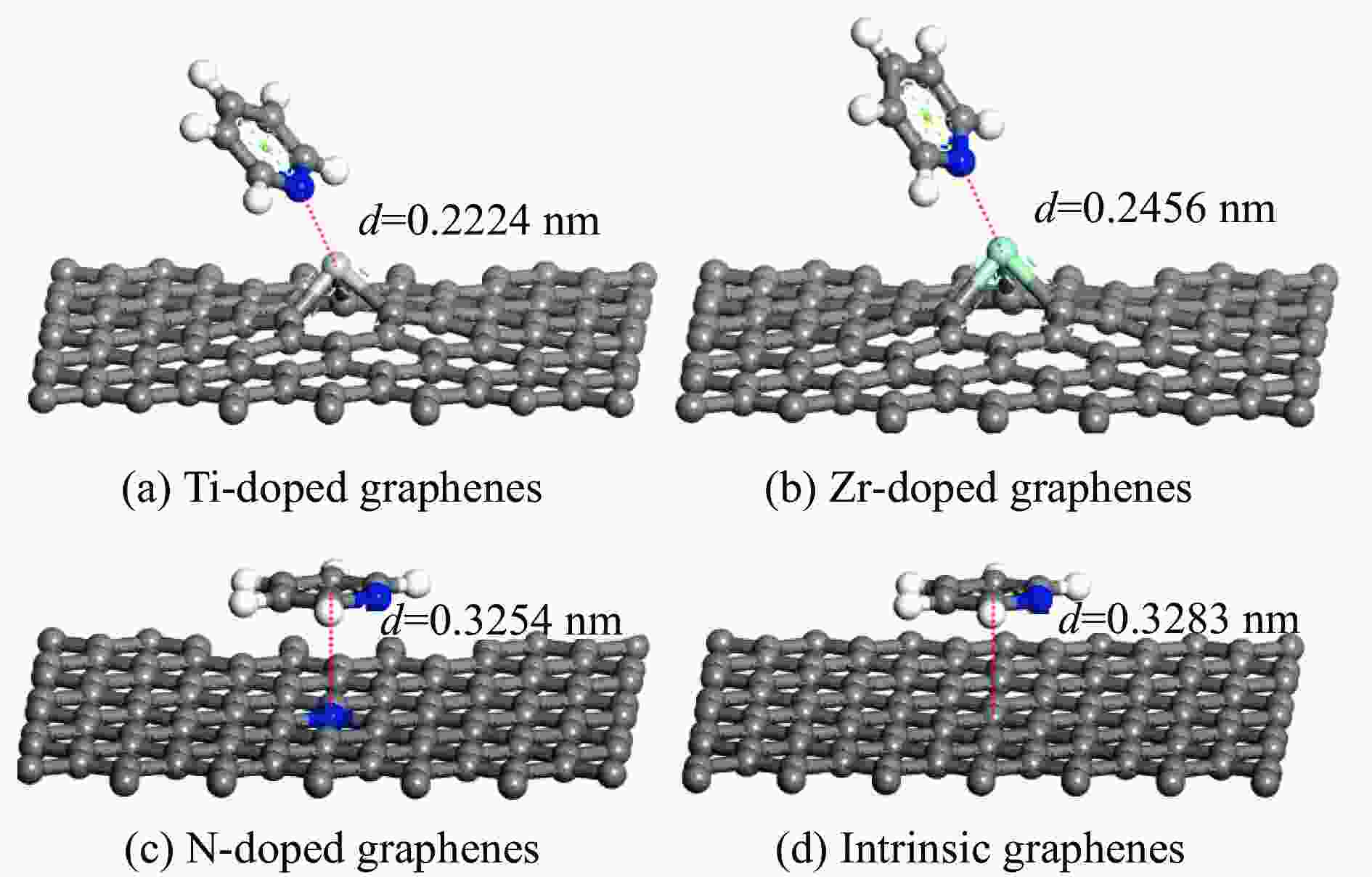
图 4 吡啶环平行吸附在各石墨烯表面的吸附构型
Figure 4 Pyridine lies adsorption on intrinsic and Ti、Zr、N doped graphenes in parallel and the ring centre of pyridine at the top of doped atom (Titanium, Zirconium, Nitrogen, carbon and hydrogen atoms are represented by incanus, green, blue, grey and white, respectively)

图 5 吡啶氮平行吸附在各石墨烯表面的吸附构型
Figure 5 Pyridine lies adsorption on intrinsic and Ti、Zr、N doped graphenes in parallel and the N of pyridine at the top of doped atom(Titanium, Zirconium, Nitrogen, carbon and hydrogen atoms are represented by incanus, green, blue, grey and white, respectively)
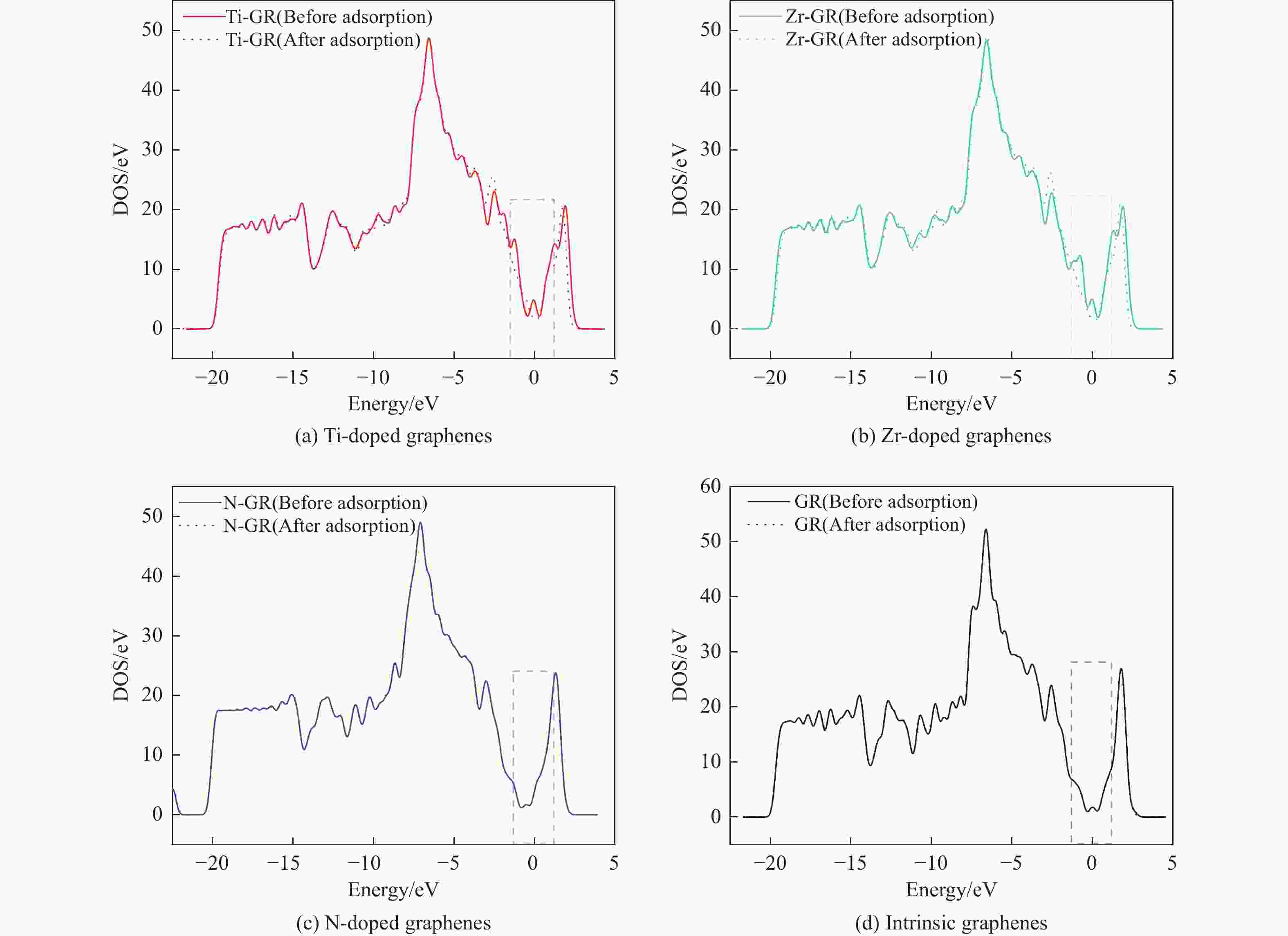
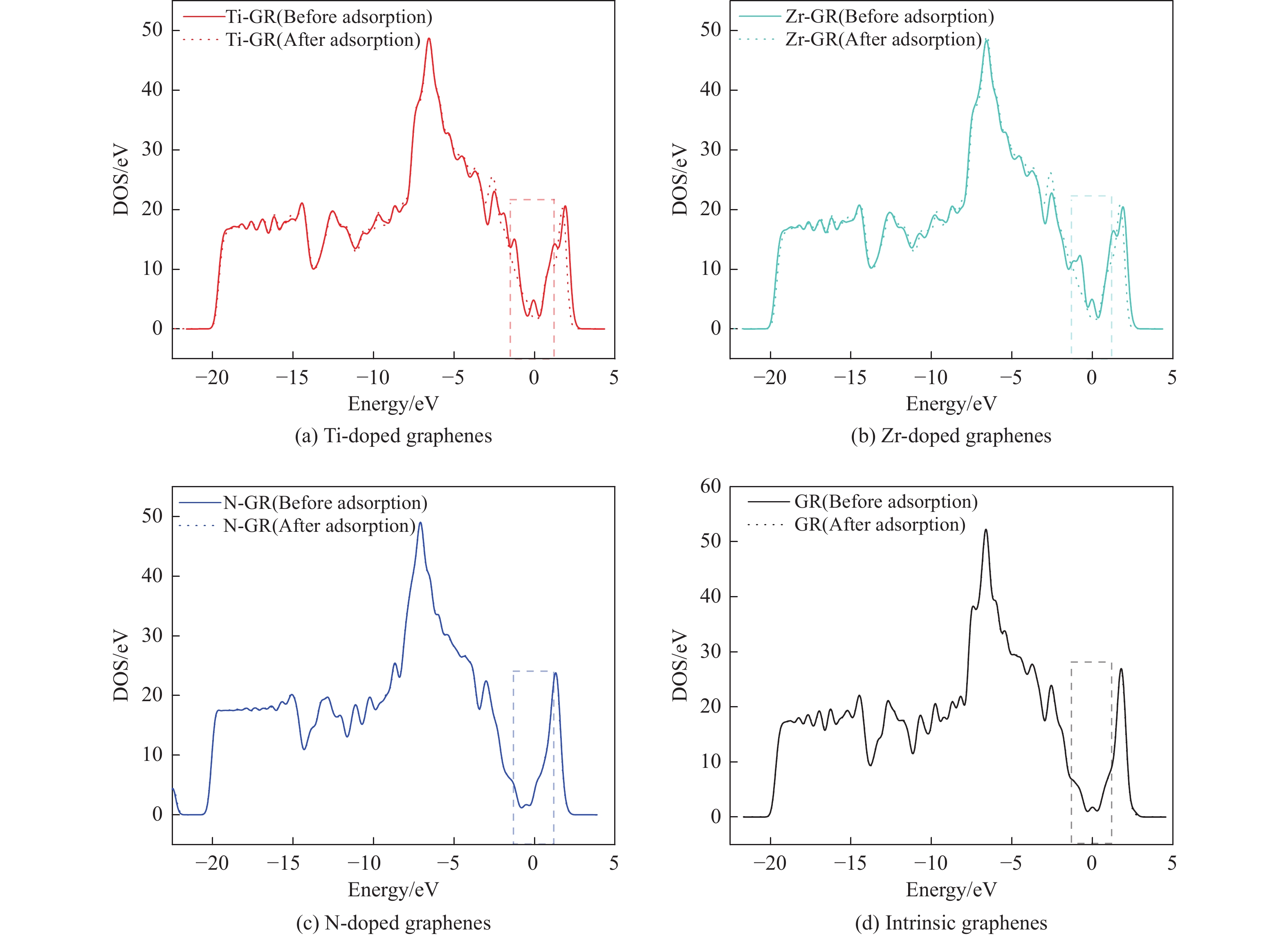
图 6 吡啶吸附在各石墨烯表面吸附前后的态密度
Figure 6 Total density of states of clean intrinsic and Ti, Zr, N doped graphenes and pyridine adsorption on intrinsic and Ti, Zr, N doped graphenes adsorption system
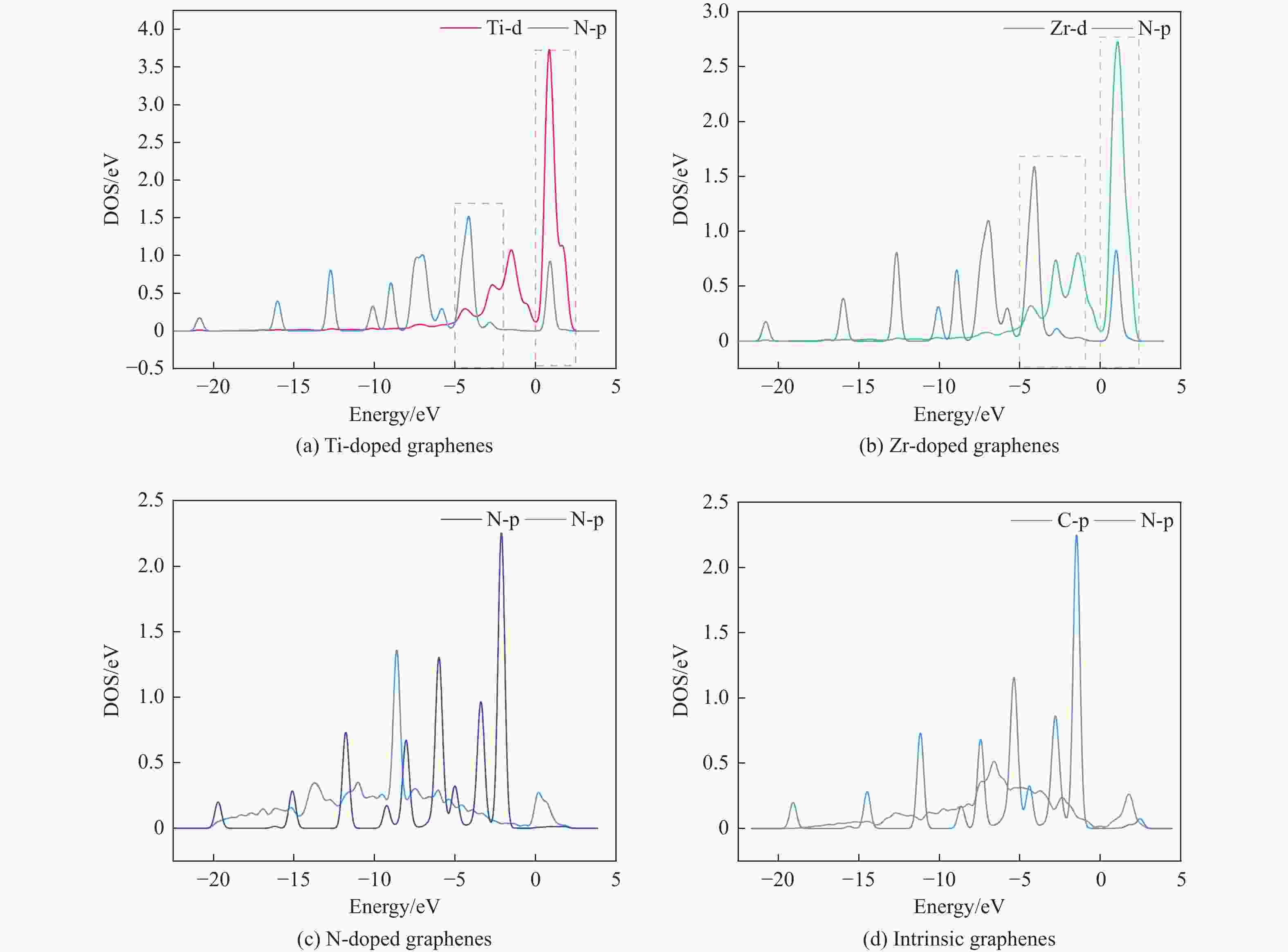
图 7 吡啶吸附在各石墨烯表面体系中吸附位点分波态密度
Figure 7 Partial density of states of the N of pyridine and Ti、Zr、N、C in adsorption system
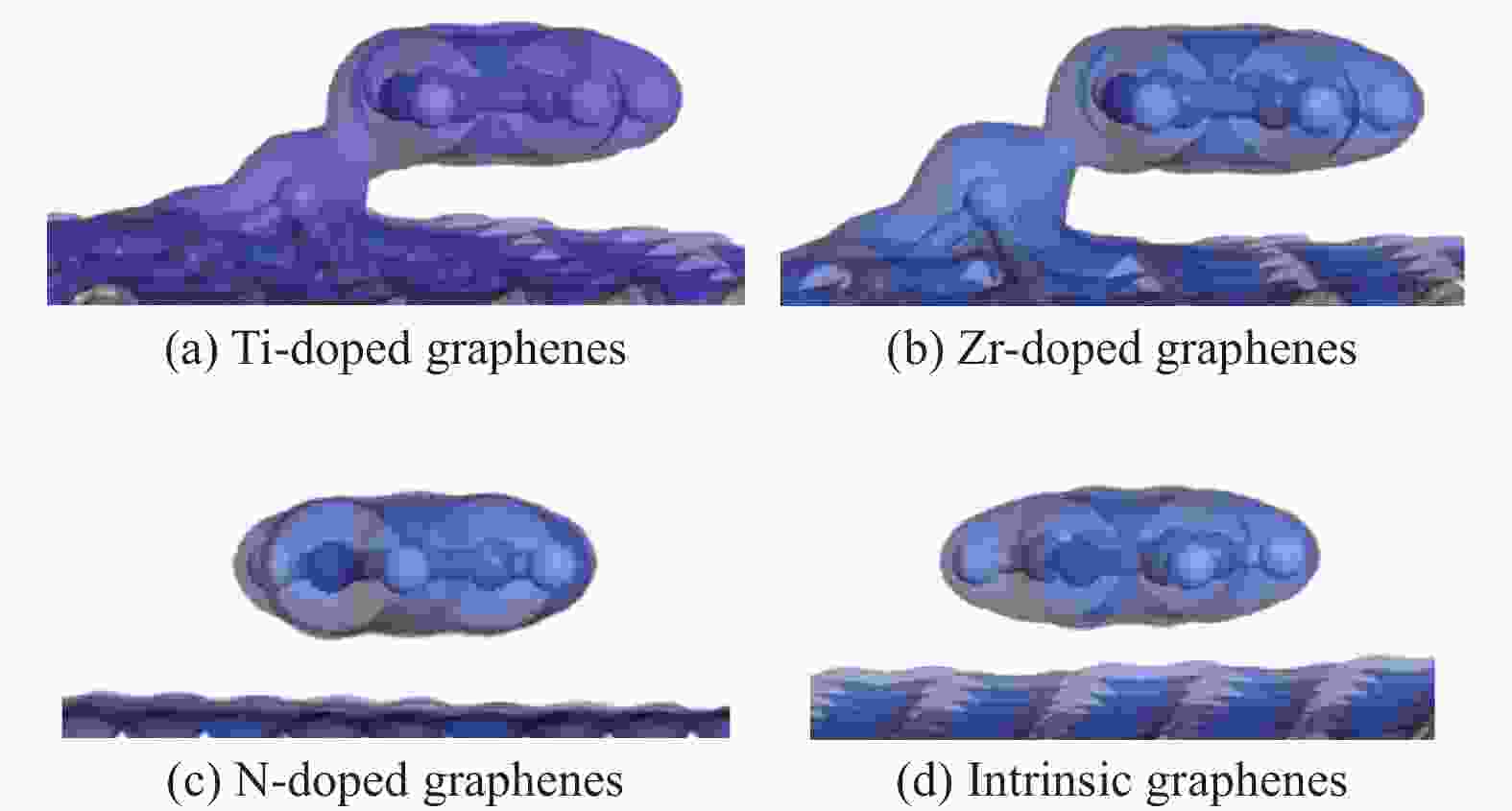
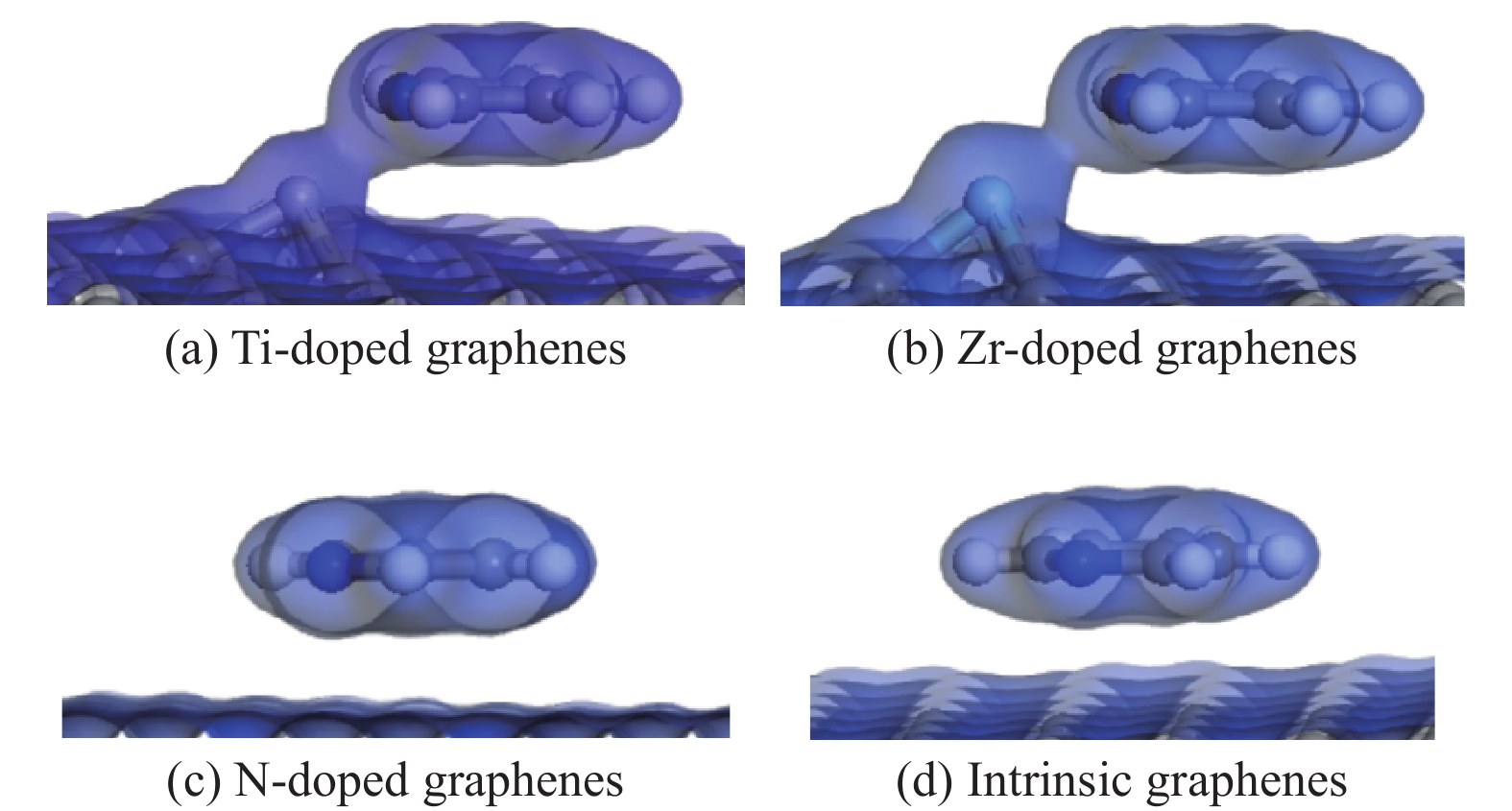
图 8 吡啶吸附在各石墨烯表面的电子密度
Figure 8 Isosurface of electron density of pyridine adsorption on intrinsic and Ti、Zr、N doped graphenes (titanium, zirconium, nitrogen, carbon and hydrogen atoms are represented by incanus, green, blue, grey and white, respectively)
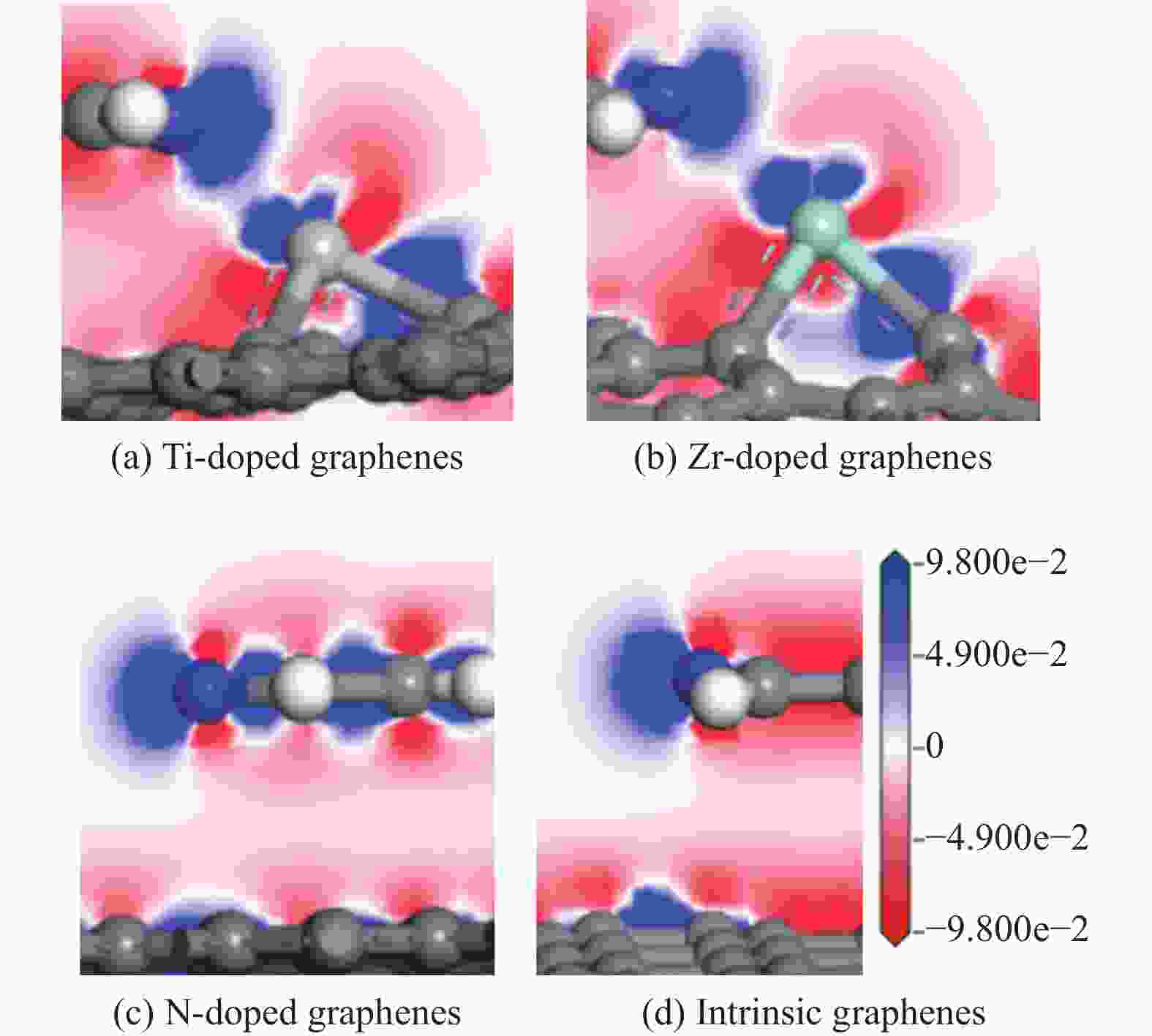
图 9 吡啶吸附在各石墨烯表面的差分电荷密度
Figure 9 Isosurface of differential charge density of pyridine adsorption on intrinsic and Ti, Zr, N doped graphenes (Titanium, Zirconium, Nitrogen, carbon and hydrogen atoms are represented by incanus, green, blue, grey and white, respectively)
表 1 各石墨烯表面的几何参数
Table 1 Geometrical parameters of the intrinsic and Ti, Zr, N doped graphenes
Doped graphenes Bond length dC-x/nm Bond angle ∠C-X-C/(°) Ti-doped graphenes 0.1775−0.1778 119.905−120.048 Zr-doped graphenes 0.1869−0.1871 119.836−120.082 N-doped graphenes 0.1409−0.1411 119.997−120.006 intrinsic graphenes 0.1420 120  下载: 导出CSV
下载: 导出CSV
表 2 吡啶在各石墨烯表面上三种吸附构型的吸附能
Table 2 Adsorption energy of pyridine adsorption on intrinsic and Ti、Zr、N doped graphenes
Adsorption structure Energy/eV Ti-doped graphenes Zr-doped graphenes N-doped graphenes Intrinsic graphenes Vertical −1.857 −1.590 −0.499 −0.374 Parallel(ring) −1.854 −1.583 −0.767 −0.736 Parallel(N) −2.034 −1.853 −0.734 −0.672
下载: 导出CSV
表 3 吡啶在各石墨烯表面吸附前后吡啶的马利肯电荷布居
Table 3 Mulliken charge of pyridine of pyridine before and after adsorption on intrinsic and Ti、Zr、N doped graphenes
Atom or mole cule Before adsorption/e After adsorption (Ti-GR)/e After adsorption (Zr-GR)/e After adsorption (N-GR)/e After adsorption (GR)/e s-orbital electron p-orbital electron Mulliken population s-orbital electron p-orbital electron Mulliken population s-orbital electron p-orbital electron Mulliken population s-orbital electron p-orbital electron Mulliken population s-orbital electron p-orbital electron Mulliken population C1 1.242 2.8 −0.042 1.236 2.819 −0.056 1.23 2.823 −0.053 1.242 2.798 −0.041 1.242 2.799 −0.04 C2 1.242 2.834 −0.076 1.237 2.865 −0.101 1.23 2.867 −0.096 1.246 2.832 −0.076 1.245 2.832 −0.08 C3 1.205 2.738 0.057 1.195 2.685 0.121 1.184 2.687 0.129 1.21 2.73 0.06 1.21 2.731 0.058 N 1.59 3.705 −0.295 1.603 3.908 −0.511 1.602 3.929 −0.531 1.59 3.732 −0.322 1.591 3.709 −0.3 C5 1.205 2.738 0.057 1.194 2.676 0.13 1.184 2.691 0.125 1.21 2.728 0.062 1.21 2.73 0.059 C6 1.242 2.834 −0.076 1.237 2.865 −0.102 1.23 2.866 −0.095 1.246 2.832 −0.076 1.245 2.832 −0.08 H7 0.923 0 0.077 0.87 0 0.13 0.876 0 0.124 0.918 0 0.082 0.921 0 0.079 H8 0.925 0 0.075 0.868 0 0.132 0.874 0 0.126 0.92 0 0.08 0.923 0 0.077 H9 0.925 0 0.075 0.859 0 0.141 0.872 0 0.128 0.917 0 0.083 0.921 0 0.079 H10 0.925 0 0.075 0.854 0 0.146 0.872 0 0.128 0.918 0 0.082 0.922 0 0.078 H11 0.925 0 0.075 0.868 0 0.132 0.874 0 0.126 0.92 0 0.08 0.923 0 0.077 py 12.349 17.649 0.002 12.021 17.818 0.162 12.03 17.863 0.111 12.337 17.652 0.014 12.353 17.633 0.01
下载: 导出CSV
表 4 吡啶在各石墨烯表面吸附前后各石墨烯的马利肯电荷布居
Table 4 Mulliken charge of intrinsic and Ti、Zr、N doped graphenes of pyridine before and after adsorption on intrinsic and Ti、Zr、N doped graphenes
Atom or
moleculeBefore adsorption/e After adsorption/e s-orbital electron p-orbital electron d-orbital electron Mulliken population s-orbital electron p-orbital electron d-orbital electron Mulliken population Ti 2.616 6.135 2.824 0.425 2.658 6.384 2.625 0.333 Zr 2.471 6.015 2.786 0.728 2.747 6.317 2.394 0.541 N 1.551 3.907 − −0.458 1.542 3.87 − −0.412 C 1.301 2.699 − 0 1.302 2.665 − 0.032 Ti-GR − − − 0 − − − −0.160 Zr-GR − − − 0 − − − −0.109 N-GR − − − 0 − − − −0.012 GR − − − 0 − − − −0.008
下载: 导出CSV
-
[1] 李玲. 燃油脱氮的可见光光催化剂制备及其性能研究[D]. 福建: 福建师范大学, 2013: 5-20.LI Ling. Visible-light photocatalysts for denitrogenation of fuel oil: preparation and characterization[D]. Fujian Normal University, 2013: 5-20.) [2] WU Ying, XIAO Jing, WU Luoming, et al. Adsorptive denitrogenation of fuel over metal organic frameworks: Effect of N-types and adsorption mechanisms[J]. J. Phys. Chem. C,2014,118(39):22533−22543. doi: 10.1021/jp5045817 [3] Ahmed I, Khan N A, Jhung S H. Adsorptive denitrogenation of model fuel by functionalized UiO-66 with acidic and basic moieties[J]. Chem. Eng. J,2017,321(1):40−47. [4] ZHANG Jun, HUANG Lei, LIN Xiongchao, et al. Effective adsorptive denitrogenation from model fuels over CeY zeolite[J]. Ind. Eng. Chem. Res,2022,61(39):14586−14597. doi: 10.1021/acs.iecr.2c01204 [5] WEN Jie, LIN Hongfei, HAN Xue, et al. Physicochemical studies of adsorptive denitrogenation by oxidized activated carbons[J]. Ind. Eng. Chem. Res,2017,56(17):5033−5041. doi: 10.1021/acs.iecr.6b05015 [6] Xin Hu, Zareen Zuhra, Shafqat Ali, et al. Adsorptive denitrogenation of model oil by MOF(Al)@GO composites: remarkable adsorption capacity and high selectivity[J]. New. J. Chem,2023,47(7):3306−3311. doi: 10.1039/D2NJ06032A [7] LI Zimeng , LIANG Hongwei , LI Xiaoling, et al. Adjusting surface acidity of hollow mesoporous carbon nanospheres for enhanced adsorptive denitrogenation of fuels[J]. Chem. Eng. Sci, 2020, 228(31): 115963. [8] Arvin Saffarian Delkhosh, Amir Vahid, Sahar Baniyaghoob, et al. Deep denitrogenation of model diesel fuel using Ni-doped mesoporous carbon: synthesis route and adsorption study[J]. ChemistrySelect.,2021,6(5):1073−1081. doi: 10.1002/slct.202004522 [9] Narjes Ghaloum, Muhieddine A. Safa, Mohammed S. Alshemali, et al. Adsorptive denitrogenation of coker diesel over carbon-based adsorbents for producing ultralow-sulfur diesel[J]. Ind. Eng. Chem. Res,2023,62(50):21777−21786. doi: 10.1021/acs.iecr.3c03424 [10] 原卫华, 毕世华, 曹茂盛. 石墨烯吸附甲醛的第一性原理研究[J]. 材料导报,2015,29(18):156−159.YUAN Weihua, BI Shihua, CAO Maosheng. Formaldehyde molecule adsorbed on graphene: a first principles study[J]. M. R,2015,29(18):156−159. [11] 罗慧娟. 石墨烯的结构掺杂及分子吸附性能研究[D]. 西北工业大学, 2017: 12-14.LUO Huijuan. Structural modifications and adsorption properties of graphene[D]. Northwestern Polytechnical University, 2017: 12-14.) [12] ZHANG Hongping, He Weidong, Luo Xuegang, et al. Adsorption of 2, 3, 7, 8-tetrochlorodibenzo-p-dioxins on intrinsic, defected, and Ti (N, Ag) doped graphene: a DFT study[J]. J. Mol. Model,2014,20(5):1−7. [13] DELLEY B. An all-electron numerical method for solving the local density functional for polyatomic molecules[J]. J. Chem. Phys,1990,92(1):508−517. doi: 10.1063/1.458452 [14] DELLEY B. From molecules to solids with the DMol3 approach[J]. J. Chem. Phys,2000,113(18):7756−7764. doi: 10.1063/1.1316015 [15] DELLEY B. DMol3 DFT studies: from molecules and molecular environments to surfaces and solids[J]. Comp. Mater. Sci,2000,17(2):122−126. [16] KRESSE G, FURTHMÜLLER J. Generalized gradient approximation made simple[J]. Phys. Rev. Lett,1996,77(18):3865−3868. doi: 10.1103/PhysRevLett.77.3865 [17] TKATCHENKO A, SCHEFFLER M. Accurate van der waals interactions from(semi)-local density functional theory[A]. APS. Meeting. Abstracts[C]. APS. Sites, 2009: 248. [18] Yoshitaka Fujimoto, Susumu Saito. Formation, stabilities, and electronic properties of nitrogen defects in graphene[J]. Phys. Rev. B, 2011, 88(24): 245446(1-7). [19] WANG Menghao, GUO Yanan, WANG Qun, et al. Density functional theory study of interactions between glycine and TiO2/graphene nanocomposites[J]. Chem. Phys. Lett,2014,599(4):86−91. [20] ZHANG Hongping, LUO Xuegang, LIN Xiaoyang, et al. Density functional theory calculations of hydrogen adsorption on Ti-, Zn-, Zr-, Al-, and N-doped and intrinsic graphene sheets[J]. Int. J. Hydrogen Energy,2013,38(33):14269−14275. doi: 10.1016/j.ijhydene.2013.07.098 [21] BEATRIZ CORDERO, VERONICA GOMEZ, ANA E, et al. Covalent radii revisited[J]. Dalton Trans,2008,37(21):2832−2838. [22] LI Jieyuan, HOU Meiling, CHEN Yanqiu, et al. Enhanced CO2 capture on graphene via N, S dual-doping[J]. Appl. Surf. Sci,2017,399(6):420−425. [23] HOU Meiling, ZHANG Xin, YUAN Shandong, et al. Double graphitic-N doping for enhanced catalytic oxidation activity of carbocatalysts[J]. Phys. Chem. Chem. Phys,2019,21(10):5481−5488. doi: 10.1039/C8CP07317A [24] DAI Jiayu, YUAN Jianmin, Giannozzi Paolo. Gas adsorption on graphene doped with B, N, Al, and S: a theoretical study[J]. Appl. Phys. Lett, 2009, 95(23): 232105(1-3). [25] CHEN Ying, LIU Yuejie, Wang Hongxia, et al. Silicon-doped graphene: an effective and metal-free catalyst for NO reduction into N2O ?[J]. ACS Appl Mater Interfaces,2013,5(13):5994−6000. doi: 10.1021/am400563g [26] CHEN Jianhoung, CHEN Hsintsung. Computational explanation for interaction between amino acid and nitrogen-containing graphene[J]. Theor. Chem. Acc,2018,137(12):176−187. doi: 10.1007/s00214-018-2392-z [27] 李巧灵, 吴晓宇, 王学伟, 等. 多孔BN选择性去除燃油中硫化合物的密度泛函理论研究[J]. 化工学报,2020,71(10):4601−4610.LI Qiaoling, WU Xiaoyu, WANG Xuewei, et al. Porous BN for selective adsorption of sulfur-containing compounds from fuel oil: DFT study[J]. CIESC Journal,2020,71(10):4601−4610. [28] SANTIAGO ALVAREZ. A cartography of the van der Waals territories[J]. Dalton Trans,2013,42(24):8617−8636. doi: 10.1039/c3dt50599e [29] 厉志鹏, 牛胜利, 赵改菊, 等. Sr掺杂对CaO(100)表面吸附甲醇影响的分子模拟[J]. 燃料化学学报,2020,48(2):172−178. doi: 10.1016/S1872-5813(20)30008-6LI Zhipeng, NIU Shengli, ZHAO Gaiju, et al. Molecular simulation study of strontium doping on the adsorption of methanol on CaO(100) surface[J]. J Fuel Chem Technol,2020,48(2):172−178. doi: 10.1016/S1872-5813(20)30008-6 [30] 张文杰, 侯美伶, 周兴, 等, 基于第一性原理计算硫化氢(H2S)在Pt-graphene上的吸附性能和解离机理[J]. 燃料化学学报, 2022, 50(9): 1211-1220.ZHANG Wenjie, HOU Meiling, ZHOU Xing, et al. A theoretical study of H2S adsorption and dissociation mechanism on defected graphene doped with Pt[J]. J Fuel Chem Technol, 2022, 50(9): 1211-1220.) [31] 王建辉, 崔建中, 王兴尧, 等. 无机化学[M]. 五版. 北京: 高等教育出版社, 2018: 334.WANG Jianhui, CUI Jianzhong, WANG Xingxiao, et al. Inorganic chemistry[M], 5nd ed, Beijing: Higher Education Press, 2018: 334.) [32] 柴汝宽, 刘月田, 杨莉, 等. 乙酸在方解石表面吸附的密度泛函研究[J]. 中南大学学报(自然科学版),2019,50(5):1252−1262.CHAI Rukuan, LIU Yuetian, YANG Li, et al. Density functional theory analysis of acetic acid adsorption on CaCO3(104) surface[J]. J. Cent. South Univ(Science and Technology),2019,50(5):1252−1262. -




 点击查看大图
点击查看大图
计量
- 文章访问数: 30
- HTML全文浏览量: 12
- PDF下载量: 7
- 被引次数: 0
